CELEBRATING 100 YEARS OF MODERN CRYSTALLOGRAPHY / CIEN AÑOS DE CRISTALOGRAFÍA MODERNA
FROM CRYSTALLOGRAPHY TO STRUCTURAL BIOLOGY, A CENTURY OF DISCOVERIES
Guillermo Montoya
University of Copenhagen
NNF Center for Protein Research
guillermo.montoya@cpr.ku.dk
ABSTRACT
From crystallography, the technique mostly used to study the structure of matter, the field mutated into structural biology, has mutated in life sciences into structural biology, which has been developed as an essential and rather successful area of research to fully understand the workings of cellular pathways. The application of physical approaches to biological systems has been crucial to comprehend the structure and function of the biological components of living organisms. In this assay the author walks the reader through the last century, which has witnessed how this life sciences research area was born and moved towards larger assemblies in the core of crucial biological problems. The influence of research in physics, biochemistry and molecular biology has been key in the successes and large body of seminal results obtained by structural biologists. The author proposes that the future of this area implies the integration of its results at the cellular level apart of using more quantitative approaches to describe biological processes.
DE LA CRISTALOGRAFÍA A LA BIOLOGÍA ESTRUCTURAL, UN SIGLO DE DESCUBRIMIENTOS
RESUMEN
La cristalografía, la técnica más ampliamente usada para estudiar la estructura de la materia, ha evolucionado en las ciencias de la vida hacia la biología estructural, una exitosa área de investigación encaminada a comprender el funcionamiento de los procesos celulares. La aplicación de aproximaciones físicas a sistemas biológicos es clave para entender la estructura y funcionamiento de los componentes de los organismos. En este artículo el autor ofrece al lector un paseo por la evolución de esta área de conocimiento durante el siglo XX, desde su nacimiento hasta el análisis de grandes complejos macromoleculares, protagonistas importantes en diversos procesos biológicos. La influencia de investigaciones en física, bioquímica y biología molecular ha sido clave para los numerosos éxitos alcanzados por biólogos estructurales. El autor sostiene que el futuro de esta disciplina pasa por la integración de sus resultados a un nivel celular además de emplear metodologías más cuantitativas para la descripción de procesos biológicos.
Received: September 12, 2014; Accepted: February 13, 2015.
Citation/Cómo citar este artículo: Montoya, G. (2015). "From Crystallography to Structural Biology, a Century of Discoveries". Arbor, 191 (772): a217. doi: http://dx.doi.org/10.3989/arbor.2015.772n2003
KEYWORDS: biophysics; crystallization; electron microscopy; macromolecular complexes; molecular biology; structural biology; X-ray crystallography.
PALABRAS CLAVE: biofísica; biología estructural; biología molecular; complejos macromoleculares; cristalización; cristalografía de rayos-X; microscopía electrónica.
Copyright: © 2015 CSIC. This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial (by-nc) Spain 3.0 License.
CONTENTS
Spectroscopy is the study of the interaction between matter and radiated energy. This physics discipline was originated through the study of light dispersed according to its wavelength by a prism. The concept was expanded to comprise any interaction with radiation as a function of its frequency. Spectroscopic data are usually represented by a spectrum, a chart showing the response of interest as a function of wavelength or frequency. Different types of radiation have been used to understand the structure of matter, but the discovery of X-rays by the German physicist Wilhelm Röntgen in 1895 (http://bit.ly/1E6Bz6U) opened the door to use this short wavelength radiation to analyze in molecular detail the structure of molecules. However, this analysis required the irradiated material in a special periodically ordered state, a crystal. Max von Laue generated the first diffraction pattern by irradiating copper sulfate crystals with X-rays in 1912 (Laue, 1913Laue, Max von (1913). "Kritische Bemerkungen zu den Deutungen der Photogramme von Friedrich und Knipping". Physikalische Zeitschrift, 14, pp. 421-423.). The same year William and Henry Bragg published the Bragg´s law, the key to decode the structural information contained in a diffraction pattern for understanding a crystal structure (http://bit.ly/1u8imBx). One year later they determined the structure of diamond. Therefore crystallography can be considered a special type of spectroscopy, whose main difference with the usual spectroscopic techniques arises from the need of crystals to obtain a “good spectrum”, the diffraction pattern.
From a physicochemical perspective crystallography made a quantum leap when the technique was applied to proteins. James Sumner had shown in the late 1920s that enzymes are proteins by crystallizing urease (The Nobel lecture: The chemical nature of enzymes, http://bit.ly/1AmuICt), and Crowfood and Bernal reported in 1934 the first X-ray diffraction of a protein crystal (Bernal & Crowfoot, 1934Bernal, J. D. and Crowfoot, D. (1934). "X-Ray photographs of crystalline pepsin". Nature, 133, pp. 794-795, http://dx.doi.org/10.1038/133794b0.). These discoveries opened the study of the molecules of life by crystallographic methods, and it can be regarded now as the true origin of the discipline now known as Structural Biology (Figure 1).
|
Figure 1. The technological pipeline of Structural Biology. Starting with protein production (1), the samples are then characterized (2) and used for Cryo-EM studies (3) and/or in the appropriated cases crystallized (4) to obtain single crystal diffraction data using synchrotron radiation (5,6) to gain atomic (7) insight into the workings of protein machines.
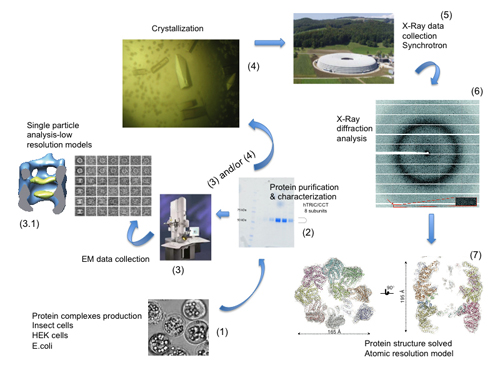
[Descargar tamaño completo]
| |
The studies on biological molecules during the following years led to many fundamental discoveries that changed completely our view of life processes. Probably one of the biggest successes was the determination of the helical structure of DNA, the molecule that carries the genetic inheritance, by Crick and Watson (Watson & Crick, 1953aWatson, J. D. and Crick, F. H. (1953a). "Molecular structure of nucleic acids, a structure for deoxyribose nucleic acid". Nature, 171, pp. 737-738, http://dx.doi.org/10.1038/171737a0.; 1953bWatson, J. D. and Crick, F. H. (1953b). "Genetical implications of the structure of deoxyribonucleic acid". Nature, 171, pp. 964-967, http://dx.doi.org/10.1038/171964b0.), based on the crystallographic work initiated by Maurice Wilkins (Wilkins et al., 1953Wilkins, M. H. F.; Stokes, A. R. and Wilson, H. R. (1953). "Molecular Structure of Nucleic Acids: Molecular Structure of Deoxypentose Nucleic Acids". Nature, 171, pp. 738-740, http://dx.doi.org/10.1038/171738a0.) and Rosalind Franklin (Franklin & Goslind, 1953Franklin, R. E. and Gosling, R. G. (1953). "Molecular Configuration of Sodium Thymonucleate". Nature, 171, pp. 740-741, http://dx.doi.org/10.1038/171740a0.). Also the crystal structures of myoglobin (Kendrew et al., 1958Kendrew, J. C.; Bodo, G.; Dintzis, H. M.; Parrish, R. G.; Wyckoff, H. and Phillips, D. C. (1958). "A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis". Nature, 181, pp. 662-666, http://dx.doi.org/10.1038/181662a0.), hemoglobin (Perutz et al., 1960Perutz, M. F.; Rossmann, M. G.; Cullis, A. F.; Muirhead, H.; Will, G. and North, A. C. T. (1960). "Structure of Hæmoglobin: A Three-Dimensional Fourier Synthesis at 5.5-Å. Resolution, Obtained by X-Ray Analysis". Nature, 185, pp. 416-422, http://dx.doi.org/10.1038/185416a0.) and lysozyme (Blake et al., 1965Blake, C. C. F.; Koenig, D. F.; Mair, G. A.; North, A. C. T.; Phillips, D. C. and Sarma, V. R. (1965). "Structure of Hen Egg-White Lysozyme: A Three-dimensional Fourier Synthesis at 2 Å Resolution". Nature, 206, pp. 757-761, http://dx.doi.org/10.1038/206757a0.) by Kendrew, Perutz and Blake further confirmed the physicochemical nature of life and the possibility to fully understand the processes where these molecules were involved. These developments together with the work of molecular biologists such as Avery, MacLeod and McCarthy, and many others, complemented the gene theory initiated by Mendel, allowing for the very first time that physicists, chemists and biochemists addressed biological processes from a qualitative and quantitative perspective beyond the phenotypic aspects that previously characterized the work of biologists. Many of these seminal works received the Nobel Prize between 1920s and the 1960s. In fact, crystallography is the discipline that has received the largest number of Nobel Prizes including the categories of Chemistry, Physics and Medicine or Physiology.
However, one of the main limitations of this relatively young research discipline arose from the required amount of purified sample to grow crystals. The pioneers were more focused on the development of this powerful technique setting up all the mathematical and physical procedures for data collection in macromolecular crystals (Figure 2). The fact that macromolecules could be crystallized posed a challenge for data collection and data processing at that time. The number of reflections obtained for a dataset of these crystals was several orders of magnitude higher when compared to the crystals of salts or other small molecules. Also reflections were weaker due to the size of a macromolecule compared to a small compound. The number of unit cells of this material in the same crystal volume was several orders of magnitude smaller. Thus protein crystals were not great amplifiers in diffraction experiments. All these limitations together with the fact that a large amount of purified sample was needed to grow crystals of biomolecules limited the attention of the pioneers to proteins that were easy to isolate in large amounts.
|
Figure 2. Harker sections of an anomalous Patterson map used to identify the positions of the Ta6Br12 clusters used to solve the phase problem and the crystal structure in Muñoz (2011Muñoz, I. G.; Yébenes, H.; Zhou, M.; Mesa, P.; Serna, M.; Park, A. Y.; Bragado-Nilsson, E.; Beloso, A.; de Cárcer, G.; Malumbres, M.; Robinson, C. V.; Valpuesta, J. M. and Montoya, G. (2011). "Crystal structure of the open conformation of the mammalian chaperonin CCT in complex with tubulin". Nature Structural & Molecular Biology, 18, pp. 14-19, http://dx.doi.org/10.1038/nsmb.1971.).
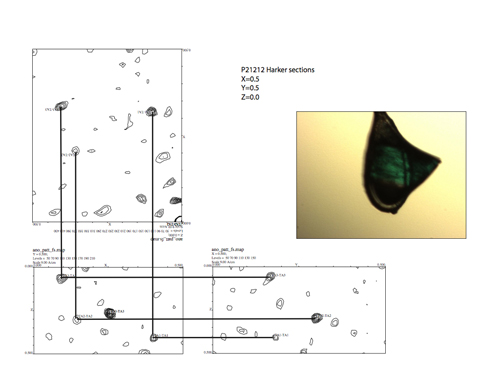
[Descargar tamaño completo]
| |
RECOMBINANT DNA ENTERS THE SCENE
Top
DNA has played an essential role in Biology and life sciences. However, probably no one predicted the strong and decisive impact that this area had in pushing Crystallography and in Structural Biology to their current levels of complexity. A key finding in the recombinant DNA technology revolution was the discovery of enzymes that could recognize and cleave DNA specifically; the isolation of these proteins in the labs of Werner Arber, Hamilton Smith and Matthew Meselson, provided the tools to edit and manipulate this nucleic acid (Roberts, 2005Roberts, R. J. (2005). "How restriction enzymes became the workhorses of molecular biology". Proceedings of the National Academy of Sciences of the USA, 102, pp. 5905-5908, http://dx.doi.org/10.1073/pnas.0500923102.). This finding together with the invention of recombinant DNA technology, the way by which genetic material from one organism is artificially introduced into the genome of another organism and then replicated and expressed by that other organism (Morrow et al., 1974Morrow, J. F.; Cohen, S. N.; Chang, A. C.; Boyer, H. W.; Goodman, H. M. and Helling, R. B. (1974). "Replication and transcription of eukaryotic DNA in Escherichia coli". Proceedings of the National Academy of Sciences of the USA, 71, pp. 1743-1747, http://dx.doi.org/10.1073/pnas.71.5.1743.) (largely the work of Paul Berg, Herbert W. Boyer, and Stanley N. Cohen), provided the way to produce large amounts of samples. With inefficient and labor-intensive purification methods and uncertain prospects for crystallization, only the most abundant and very stable medium-size proteins, such as those found in body fluids or muscles, were considered as feasible targets. The whole area might have stalled prematurely. However, shortly after the first successful expression of recombinant proteins in Escherichia coli, including insulin (Goeddel et al., 1979Goeddel, D. V.; Kleid, D. G.; Bolivar, F.; Heyneker, H. L.; Yansura, D. G.; Crea, R.; Hirose, T.; Kraszewski, A.; Itakura, K. and Riggs, A. D. (1979). "Expression in Escherichia coli of chemically synthesized genes for human insulin". Proceedings of the National Academy of Sciences of the USA, 76, pp. 106-110, http://dx.doi.org/10.1073/pnas.76.1.106.) and somatostatin (Itakura et al., 1977Itakura, K.; Hirose, T.; Crea, R.; Riggs, A. D.; Heyneker, H. L.; Bolivar, F. and Boyer, H. W. (1977). "Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin". Science, 198, pp. 1056-1063, http://dx.doi.org/10.1126/science.412251.), protein crystallographers turned to recombinant methods as the means to obtain samples for crystallization. Among the very first proteins crystallized using recombinant samples were insulin (Chance et al., 1981Chance, R. E.; Kroeff, E. P.; Hoffmann, J. A. and Frank, B. H. (1981). "Chemical, physical, and biologic properties of biosynthetic human insulin". Diabetes Care, 4, pp. 147-154, http://dx.doi.org/10.2337/diacare.4.2.147.), human leukocyte interferon A (Miller et al., 1981Miller, D. L.; Kung, H. F.; Staehelin, T. and Pestka, S. (1981). "The crystallization of recombinant human leukocyte interferon A". Methods Enzymology, 79, pp. 3-7, http://dx.doi.org/10.1016/S0076-6879(81)79005-0., 1982Miller, D. L.; Kung, H. F. and Pestka, S. (1982). "Crystallization of recombinant human leukocyte interferon A". Science, 215, pp. 689-690, http://dx.doi.org/10.1126/science.6173922.), murine interferon β (Matsuda et al., 1986Matsuda, S.; Kawano, G.; Itoh, S.; Mitsui, Y. and Iitaka, Y. (1986). "Crystallization and preliminary X-ray studies of recombinant murine interferon-β.". Journal of Biological Chemistry, 261, pp. 16207-16209.), and eglin C (Grutter et al., 1985Grutter, M. G.; Marki, W. and Walliser, H. P. (1985). "Crystals of the complex between recombinant N-acetyleglin c and subtilisin". Journal of Biological Chemistry, 260, pp. 11436-11437.), Also, recombinant methods made it feasible both the ad hoc modification of protein sequences and the use of orthologues as variables in the crystallization experiments. Prior to that point, the only way to use the protein as a variable was to screen homologues from various species. The use of purification tags based on different affinity interactions (Strep-Tag, His-Tag, GST-tag, and so forth) has also facilitated and speed-up and the isolation of proteins expressed in low amount or unstable, thus providing another way of overcoming limitations arising from protein amount and stability.
POLYMERASE CHAIN REACTION (PCR) BOOSTS STRUCTURAL BIOLOGY
Top
However, recombinant DNA techniques were not easy to use and cloning could be cumbersome at that time, limiting the throughput and the targets that were addressed by structural biologists. It was in 1983 when Kary Mullis, at that time a scientist at the Cetus Corporation, conceived PCR as a method to copy DNA and synthesize large amounts of a specific target DNA (Mullis et al., 1987Mullis, K. B.; Erlich, H. A.; Arnheim, N.; Horn, G. T.; Saiki, R. K. and Scharf, S. J. (1987). "Process for amplifying, detecting, and/or-cloning nucleic acid sequences". .; Mullis, 1990). Over the next two years, a team of Cetus scientists that recognized the potential impact of PCR could have on molecular biology, researched, refined and made the theoretical process a reality. This finding was essential for molecular biologists because it opened many possibilities for gene cloning and also facilitated the rapid sequencing of the genome of any organism. PCR has been essential in Structural Biology, not only to target many different proteins for structural studies, also because it opened the door to site-directed mutagenesis. This technique has been essential to move from pure structural studies to structure-function analysis, opening the avenues for mechanistic studies of different biological processes.
SYNCHROTRON RADIATION
Top
As previously mentioned, the fact that macromolecular crystals are fragile, small and their unit cells are large compared to small molecules imposed hard technical restrictions when irradiating them with conventional X-ray sources. As a result of these features, reflections were weak and difficult to record and the resolution was quite limited. Therefore the absence of high-brilliance sources of X-ray radiation made the crucial diffraction measurements extremely slow or impossible in some cases. A breakthrough in this topic came from the physics side (reviewed by G. Fox in this monograph). The use of synchrotrons in the 1970s represented a qualitative improvement that changed the face of macromolecular crystallography from that moment on. This ‘quantum leap’ was largely due to the introduction of X-ray radiation from synchrotron sources. The increase in X-ray flux in comparison with rotating anodes allowed many different new strategies for data collection. High quality diffraction patterns can be obtained from smaller crystals and different regions of big crystals can be sampled to obtain better data. The crystal freezing techniques, which started to be developed in the late 1960s, were also indispensable to avoid crystal decay in this strong radiation sources.
Recently the development of automatic data collection techniques, robotic systems and the new pixel array detectors in combination with these high flux sources have changed completely our data collection strategies. I remember that my first MAD data collection experiment took 3-4 days in 1994 at the ESRF. The last one we performed at the SLS was completed in 30 minutes and we were slow (Figure 3).
|
Figure 3. Detail of the sample position in one of the Swiss Light Source beamlines. The use of kappa goniometers, such as PriGo is coming back due to their possibilities to improve efficient data collection of crystals in order to avoid radiation damage

[Descargar tamaño completo]
| |
Perhaps one of the most exciting recent technical developments in the field relates to the application of X-ray free electron lasers (XFELs) to macromolecular crystallography. XFELs provide extremely intense X-ray pulses of ~ 1012 photons and ~ 40 fs in duration focused to a spot of 0.1 to 1 μm2. Since the Henderson radiation damage limit is exceeded within a single pulse, the sample rapidly disintegrates. Despite this damage process, diffraction data is collected before the destruction of the sample (Chapman et al., 2011Chapman, H. N.; Fromme, P.; Barty, A.; White, T. H.; Kirian, R. A.; Aquila, A.; Hunter, M. S.; Schulz, J.; DePonte, D. P.; Weierstall, U.; Doak, R. B.; Maia, F. R. N. C.; Martin, A. V.; Schlichting, I.; Lomb, L.; Coppola, N.; Shoeman, R. L.; Epp, S. W.; Hartmann, R.; Rolles, D.; Rudenko, A.; Foucar, L.; Kimmel, N.; Weidenspointner, G.; Holl, P.; Liang, M.; Barthelmess, M.; Caleman, C.; Boutet, S.; Bogan, M. J.; Krzywinski, J.; Bostedt, C.; Bajt, S.; Gumprecht, L.; Rudek, B.; Erk, B.; Schmidt, C.; Hömke, A.; Reich, C.; Pietschner, D.; Strüder, L.; Hauser, H.; Gorke, H.; Ullrich, J.; Herrmann, S.; Schaller, G.; Schopper, F.; Soltau, H.; Kühnel, K. U.; Messerschmidt, M.; Bozek, J. D.; Hau-Riege, S. P.; Frank, M.; Hampton, C. Y.; Sierra, R. G.; Starodub, D.; Williams, G. J.; Hajdu, J.; Timneanu, N.; Seibert, M. M.; Andreasson, J.; Rocker, A.; Jönsson, O.; Svenda, M.; Stern, S.; Nass, K.; Andritschke, R.; Schröter, C. D.; Krasniqi, F.; Bott, M.; Schmidt, K. E.; Wang, X.; Grotjohann, I.; Holton, J. M.; Barends, T. R. M.; Neutze, R.; Marchesini, S.; Fromme, R.; Schorb, S.; Rupp, D.; Adolph, M.; Gorkhover, T.; Andersson, I.; Hirsemann, H.; Potdevin, G.; Graafsma, H.; Nilsson, B. and Spence, J. C. H. (2011). "Femtosecond X-ray protein nanocrystallography". Nature, 470, pp. 73-77, http://dx.doi.org/10.1038/nature09750.) and high-resolution crystallographic data sets can be recovered by merging diffraction data from thousands of microcrystals (Johansson et al., 2012Johansson, L. C.; Arnlund, D.; White, T. A.; Katona, G.; DePonte, D. P.; Weierstall, U.; Doak, R. B.; Shoeman, R. L.; Lomb, L.; Malmerberg, E.; Davidsson, J.; Nass, K.; Liang, M.; Andreasson, J.; Aquila, A.; Bajt, S.; Barthelmess, M.; Barty, A.; Bogan, M. J.; Bostedt, C.; Bozek, J. D.; Caleman, C.; Coffee, R.; Coppola, N.; Ekeberg, T.; Epp, S. W.; Erk, B.; Fleckenstein, H.; Foucar, L.; Graafsma, H.; Gumprecht, L.; Hajdu, J.; Hampton, C. Y.; Hartmann, R.; Hartmann, A.; Hauser, G.; Hirsemann, H.; Holl, P.; Hunter, M. S.; Kassemeyer, S.; Kimmel, N.; Kirian, R. A.; Maia, F. R. N. C.; Marchesini, S.; Martin, A. V.; Reich, C.; Rolles, D.; Rudek, B.; Rudenko, A.; Schlichting, I.; Schulz, J.; Seibert, M. M.; Sierra, R. G.; Soltau, H.; Starodub, D.; Stellato, F.; Stern, S.; Strüder, L.; Timneanu, N.; Ullrich, J.; Wahlgren, W. Y.; Wang, X.; Weidenspointner, G.; Wunderer, C.; Fromme, P.; Chapman, H. N.; Spence, J. C. H. and Neutze, R. (2012). "Lipidic phase membrane protein serial femtosecond crystallography". Nature Methods, 9, pp. 263-265, http://dx.doi.org/10.1038/nmeth.1867.). Since every crystal exposed to the XFEL beam yields only a single diffraction image, data is collected from a continuous flow of microcrystals and the approach has been coined serial crystallography. As well as facilitating the collection of diffraction data from nanocrystals, serial crystallography is a room temperature approach and allows time-resolved studies to be pursued.
THE RISE OF STRUCTURE
Top
By the end of the 1990s the basis of Structural Biology were solidly placed, 30-40 years after the pioneering work of Perutz and Kendrew. Structural Biology became, and still is, a crossroad between biochemistry, molecular biology, biophysics, crystallography and cellular biology. Altogether these different subjects played, and will play, very important roles in the development of many structural projects, representing a challenge for scientists.
In 1999 a perspective article by Sali & Kuriyan predicted the future of Structural Biology during the following 15 years (Sali & Kuriyan, 1999Sali, A. and Kuriyan, J. (1999). "Challenges at the frontiers of structural biology". Trends in Cell Biology, 9, pp. M20-M24, http://dx.doi.org/10.1016/S0962-8924(99)01685-2.). In that paper the authors argue that to completely understand the molecular mechanisms of cellular processes a detailed knowledge of the structures of all cellular components at an atomic level would be necessary. The progress of this research expanded the frontiers of structural biology in two different directions. All the different advances in cloning, protein expression, purification, crystallization and synchrotron data collection prompted the generation of consortia aiming to solve all the structures of an organism proteome. This new area termed ‘structural genomics’ was very much inspired by the growing impact of genome-sequencing efforts. The other research line seeks to analyse the structures of complex molecular assemblies that are ever larger and more intricate.
STRUCTURAL GENOMICS
Top
Different consortia were aimed at accelerating the rate of solving protein structures to discover new folds. Several consortia in the US and Europe have been rather successful in this objective. Since the initial paper reporting the crystallization and the structure solution of a large percentage of the proteome of Thermotoga maritima (Lesley et al., 2002Lesley, S. A.; Kuhn, P.; Godzik, A.; Deacon, A. M.; Mathews, I.; Kreusch, A.; Spraggon, G.; Klock, H. E.; McMullan, D.; Shin, T.; Vincent, J.; Robb, A.; Brinen, L. S.; Miller, M. D.; McPhillips, T. M.; Miller, M. A.; Scheibe, D.; Canaves, J. M.; Guda, C.; Jaroszewski, L.; Selby, T. L.; Elsliger, M. A.; Wooley, J.; Taylor, S. S.; Hodgson, K. O.; Wilson, I. A.; Schultz, P. G. and Stevens, R. C. (2002). "Structural genomics of the Thermotoga maritime proteome implemented in a high-throughput structure determination pipeline". Proceedings of the National Academy of Sciences of the USA, 99, pp. 11664-11669, http://dx.doi.org/10.1073/pnas.142413399.) till recent advances in the study of kinase and protein binders of epigenetic protein binders (Structural Genomics Consortia in Oxford http://bit.ly/1Anc39I and Toronto http://bit.ly/1CvucF1), this approach has been extremely useful to complete the protein folding universe. In fact, in the last 4-5 years there has not been a protein structure deposition in the PDB that depicts a new fold. This could indicate that the structure sampling contained in the PDB is ample enough and few folds, if any, are missing. On the other hand it could also suggest that we have collected the “low-hanging” fruit of the “protein folding tree”, implying that if we want to reach the upper fruit new technology and extra efforts would be necessary. This latter option is supported by the fact that the success ratio of these consortia from gene to structure is relatively low, and this ratio is even lower when the targets are from eukaryotic origin. Nevertheless, these consortia have developed new technologies, including from automation and miniaturization of protein expression and crystallization up to synchrotron data collection, technologies that have been further employed in solving more complicated projects. An example of this are the slow but steady flow of large protein complexes structures, which represent another quantum leap for the understanding of basic processes, such as chromatin structure (Makde et al., 2010Makde, R. D.; England, J. R.; Yennawar, H. P. and Tan, S. (2010). "Structure of RCC1 chromatin factor bound to the nucleosome core particle". Nature, 467, pp. 562-566, http://dx.doi.org/10.1038/nature09321.), protein translation (Ben-Shem et al., 2011Ben-Shem, A.; Garreau de Loubresse, N.; Melnikov, S.; Jenner, L.; Yusupova, G. and Yusupov, M. (2011). "The structure of the eukaryotic ribosome at 3.0 Å resolution". Science, 334, pp. 1524-1529, http://dx.doi.org/10.1126/science.1212642.), transcription (Cramer et al., 2000Cramer, P.; Bushnell, D. A.; Fu, J.; Gnatt, A. L.; Maier-Davis, B.; Thompson, N. E.; Burgess, R. R.; Edwards, A. M.; David, P. R. and Kornberg, R. D. (2000). "Architecture of RNA polymerase II and implications for the transcription mechanism". Science, 288, pp. 640-649, http://dx.doi.org/10.1126/science.288.5466.640.), splicing (Pomeranz-Krummel et al., 2009Pomeranz-Krummel, D. A.; Oubridge, C.; Leung, A. K.; Li, J. and Nagai, K. (2009). "Crystal structure of human spliceosomal U1 snRNP at 5.5 Å resolution". Nature, 458, pp. 475-480, http://dx.doi.org/10.1038/nature07851.), protein folding (Muñoz et al., 2011Muñoz, I. G.; Yébenes, H.; Zhou, M.; Mesa, P.; Serna, M.; Park, A. Y.; Bragado-Nilsson, E.; Beloso, A.; de Cárcer, G.; Malumbres, M.; Robinson, C. V.; Valpuesta, J. M. and Montoya, G. (2011). "Crystal structure of the open conformation of the mammalian chaperonin CCT in complex with tubulin". Nature Structural & Molecular Biology, 18, pp. 14-19, http://dx.doi.org/10.1038/nsmb.1971.), protein degradation (Śledź et al., 2013Śledź, P.; Unverdorben, P.; Beck, F.; Pfeifer, G.; Schweitzer, A.; Förster, F. and Baumeister, W. (2013). "Structure of the 26S proteasome with ATP-γS bound provides insights into the mechanism of nucleotide-dependent substrate translocation". Proceedings of the National Academy of Sciences of the USA, 110, pp. 7264-7269, http://dx.doi.org/10.1073/pnas.1305782110.), DNA repair (Sibanda et al., 2010Sibanda, B. L.; Chirgadze, D. Y. and Blundell, T. L. (2010). "Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats". Nature, 463, pp. 118-121, http://dx.doi.org/10.1038/nature08648.) and many more.
MACROMOLECULAR ASSEMBLIES
Top
The original motivation for crystallizing proteins was to purify a specific macromolecule from a complex extract, or to demonstrate, in the classical chemist’s sense, the homogeneity of a preparation. Throughout that period, crystallinity was associated with purity. In the late 1930s, Astbury, Bernal, Crowfoot, Kendrew, and Perutz, turned their attention to protein crystals as a source of structural information. Their seminal studies always used samples isolated from rich natural sources, for example a specialized organ producing high amounts of a given protein or an organism, which is adapted to a certain environment requiring a specialized protein system. This strategy has been constantly employed since for the isolation of molecular machines later subjected to structural analysis. Structures of endogenously purified samples are largely restricted to molecular machines with basic cellular functions in transcription (RNA polymerase II) (Edwards et al., 1990Edwards, A. M.; Darst, S. A.; Feaver, W. J.; Thompson, N. E.; Burgess, R. R. and Kornberg, R. D. (1990). "Purification and lipid-layer crystallization of yeast RNA polymerase II". Proceedings of the National Academy of Sciences of the USA, 87, pp. 2122-2126, http://dx.doi.org/10.1073/pnas.87.6.2122.), translation (ribosome) (Clemons et al., 2001Clemons, W. M. Jr.; Brodersen, D. E.; McCutcheon, J. P.; May, J. L.; Carter, A. P.; Morgan-Warren, R. J.; Wimberly, B. T. and Ramakrishnan, V. (2001). "Crystal structure of the 30 S ribosomal subunit from Thermus thermophilus: purification, crystallization and structure determination". Journal of Molecular Biology, 310, pp. 827-843, http://dx.doi.org/10.1006/jmbi.2001.4778.), transport (photosynthetic and respiratory chain complexes) (Jordan et al., 2001Jordan, P.; Fromme, P.; Witt, H. T.; Klukas, O.; Saenger, W. and Krauss, N. (2001). "Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution". Nature, 411, pp. 909-917, http://dx.doi.org/10.1038/35082000.) and energy metabolism (fatty acid synthase) (Maier et al., 2006Maier, T.; Jenni, S. and Ban, N. (2006). "Architecture of mammalian fatty acid synthase at 4.5 Å resolution". Science, 311, pp. 1258-1262, http://dx.doi.org/10.1126/science.1123248.). Although recombinant expression techniques are constantly evolving, there are still key biological processes regulated by proteins or protein complexes, which are difficult to obtain by recombinant methods. Therefore the use of endogenous preparations for the study of macromolecular machines will be always an option, which is currently used with regular frequency.
However, crystals of macromolecular complexes are often difficult to obtain and their diffraction is usually weak due to large unit cells and high solvent content. This combination together with their usually reduced size makes the crystals of these specimens rather difficult to manipulate. Another problem arises from the fact that macromolecular machines isolated from a natural source are generally heavily modified. The different subunits can contain multiple posttranslational modifications, which introduce an extra source of heterogeneity that could affect the quality of the crystals or even hinder crystallization. Therefore the development of new strategies for the structural analysis of large complexes has been one of the main research areas in the field during the last years. The main development arises from the use of co-expression systems to obtain the “in cell” assembly of the protein complex. Although coexpression in E.coli was performed with homemade systems using intelligent strategies with restriction enzymes, the recent introduction of the multibac method using insect cells was a big success for the study of protein complexes (Berger et al., 2004Berger, I.; Fitzgerald, D. J. and Richmond, T. J. (2004). "Baculovirus expression system for heterologous multiprotein complexes". Nature Biotechnology, 22, pp. 1583-1587, http://dx.doi.org/10.1038/nbt1036.). This method allows the use of a single baculovirus for the coexpression of the multiple components of the protein complex. The system has been extended for homologous or heterologous expression to mammalian cells and E.coli. The use of these systems expands the number hosts and provides a wider range to address the different problems that can be faced by the researcher. The generation of recombinant samples is essential for the success of this type of projects, not only for crystallization but also for further structure-function analysis.
The use of the new third-generation synchrotron radiation sources and X-FEL is fundamental to collect data from these, normally, weakly diffracting crystals. The development of automatic sample mounting systems and the use of this high flux beams facilitate data collection. However, these powerful X-ray sources cause substantial radiation damage on the samples, which can be alleviated using multiple crystals for data collection. The development of a new generation of X-ray detectors based on single photon counting pixel arrays is another factor that has greatly enhanced the quality of the data collected from this type of crystals (Mueller et al., 2012Mueller, M.; Wang, M. and Schulze-Briese, C. (2012). "Optimal fine Φ-slicing for single-photon-counting pixel detectors". Acta Crystallographica D Biological Crystallography, 68, pp. 42-56, http://dx.doi.org/10.1107/S0907444911049833.). These detectors have radically transformed X-ray research at synchrotron beam lines and in laboratory applications. Their unique properties enable improved data acquisition protocols or even completely new experiments, resulting in higher throughput, provoking a paradigm shift in the way experiments are done (continuous, shutter-less mode). Therefore the combination of new molecular biology approaches together with further technological developments has elevated Structural Biology projects to a higher level of complexity.
ELECTRON MICROSCOPY
Top
Viruses and large macromolecular assemblies have been observed using electron microscopy for a long time, but during the last 30 years this field has suffered a big transformation, and for the first time the use of single particle averaging from electron microcopy micrographs has begun to provide models of large molecular complexes at a resolution comparable to those solved using crystallographic methods. The use of the new direct electron detectors, which are photon-counting devices similar to those use for X-rays that have been commented previously (McMullan et al., 2009McMullan, G.; Chen, S.; Henderson, R. and Faruqi, A. R. (2009). "Detective quantum efficiency of electron area detectors in electron microscopy". Ultramicroscopy, 109, pp. 1126-1143, http://dx.doi.org/10.1016/j.ultramic.2009.04.002.), has revolutionized the field making possible for certain samples the dream of crystallography without crystals. A fundamental principle of crystallography resides in the use of redundancy to achieve a virtually noise-free average. This almost noise-free averaging has become possible in EM due to the development of the new detectors, which provide high signal-to-noise ratio for the images of cryo preserved specimens. Although the level of damage caused by the electron beam is quite large, the use of this type of detectors has also decreased the number of particles that are needed to achieve a proper sample analysis. The combination of these new advancements with classical crystallographic studies and improved modeling approaches could initiate a new era, providing researchers with the appropriate structural scenario to test different hypothesis of key cellular processes.
AND, WHAT ABOUT THE FUTURE…?
Top
In this article I was trying to walk the reader from the beginnings to the current status of Structural Biology, but where are we going? How the field will develop in the future 20-40 years? Although biologists are starting to understand many of the secrets of genome regulation and cell fate, this knowledge is normally qualitative. This hinders in many cases the possibility to make and fulfill predictions. In my opinion biology, and in particular cellular and molecular biology, face the challenge of becoming exact sciences in the following decades. While physicists are able to send an object to Mars or predict the existence of subatomic particles, molecular biologists are still not able to predict a drug target or certain phenotypes when acting over a subset of genes. Why? What is the difference? In my opinion the difference arises from the fact that physicists have a qualitative and quantitative knowledge of the mechanisms involved in these processes, while molecular and cellular biologists do not. Molecular biologists have a good, although not complete, qualitative knowledge of many important cellular pathways, but we do not know how any molecules of a certain kinase are needed to phosphorylate its target and trigger a signaling cascade, or how many molecules of growth factor are needed to fully activate the receptor downstream response. Structural Biology should play a fundamental role in this type of analysis; however, new approaches and methods must be developed if we want to accomplish such a level of detail. The confluence of structural biology with a more systematic and quantitative way of study of cellular process, the so-called systems biology area, will be essential if we want to start to make detailed predictions to test our hypothesis. The combination of all these methods will develop the future Structural Biology area into a hybrid discipline where researchers will need to gather and combine different expertise to address complex questions. Similarly to the old times the solution will come from the interplay between different research areas. As Francis Crick stated 25 years ago “I think that one should approach these problems at all levels, as was done in molecular biology. Classical genetics is, after all, a black-box subject. The important thing was to combine it with biochemistry. In nature hybrid species are usually sterile, but in science the reverse is often true. Hybrid subjects are often astonishingly fertile, whereas if a scientific discipline remains too pure it usually wilts.”
ACKOWLEDGEMENTSTop
I want to thank the Spanish Government (1990-1993), EMBO, The EU Marie Curie Program and The Peter und Traudl Engelhorn Foundation for supporting my Ph.D. and postdoctoral education. I also want to thank my Ph.D. supervisor, Rafael Picorel, for transmitting me his strong commitment with research. My work is currently supported by the Ministerio de Economía y Competitividad [BFU2011-23815/BMC, the Novo Nordisk Foundation Grant NNF14CC0001, the Fundación Ramón Areces and the Comunidad Autónoma de Madrid CAM-S2010/BMD-2305.
REFERENCESTop
|
| ○ | Ben-Shem, A.; Garreau de Loubresse, N.; Melnikov, S.; Jenner, L.; Yusupova, G. and Yusupov, M. (2011). "The structure of the eukaryotic ribosome at 3.0 Å resolution". Science, 334, pp. 1524-1529. http://dx.doi.org/10.1126/science.1212642 |
| ○ | Berger, I.; Fitzgerald, D. J. and Richmond, T. J. (2004). "Baculovirus expression system for heterologous multiprotein complexes". Nature Biotechnology, 22, pp. 1583-1587. http://dx.doi.org/10.1038/nbt1036 |
| ○ | Bernal, J. D. and Crowfoot, D. (1934). "X-Ray photographs of crystalline pepsin". Nature, 133, pp. 794-795. http://dx.doi.org/10.1038/133794b0 |
| ○ | Blake, C. C. F.; Koenig, D. F.; Mair, G. A.; North, A. C. T.; Phillips, D. C. and Sarma, V. R. (1965). "Structure of Hen Egg-White Lysozyme: A Three-dimensional Fourier Synthesis at 2 Å Resolution". Nature, 206, pp. 757-761. http://dx.doi.org/10.1038/206757a0 |
| ○ | Chance, R. E.; Kroeff, E. P.; Hoffmann, J. A. and Frank, B. H. (1981). "Chemical, physical, and biologic properties of biosynthetic human insulin". Diabetes Care, 4, pp. 147-154. http://dx.doi.org/10.2337/diacare.4.2.147 |
| ○ | Chapman, H. N.; Fromme, P.; Barty, A.; White, T. H.; Kirian, R. A.; Aquila, A.; Hunter, M. S.; Schulz, J.; DePonte, D. P.; Weierstall, U.; Doak, R. B.; Maia, F. R. N. C.; Martin, A. V.; Schlichting, I.; Lomb, L.; Coppola, N.; Shoeman, R. L.; Epp, S. W.; Hartmann, R.; Rolles, D.; Rudenko, A.; Foucar, L.; Kimmel, N.; Weidenspointner, G.; Holl, P.; Liang, M.; Barthelmess, M.; Caleman, C.; Boutet, S.; Bogan, M. J.; Krzywinski, J.; Bostedt, C.; Bajt, S.; Gumprecht, L.; Rudek, B.; Erk, B.; Schmidt, C.; Hömke, A.; Reich, C.; Pietschner, D.; Strüder, L.; Hauser, H.; Gorke, H.; Ullrich, J.; Herrmann, S.; Schaller, G.; Schopper, F.; Soltau, H.; Kühnel, K. U.; Messerschmidt, M.; Bozek, J. D.; Hau-Riege, S. P.; Frank, M.; Hampton, C. Y.; Sierra, R. G.; Starodub, D.; Williams, G. J.; Hajdu, J.; Timneanu, N.; Seibert, M. M.; Andreasson, J.; Rocker, A.; Jönsson, O.; Svenda, M.; Stern, S.; Nass, K.; Andritschke, R.; Schröter, C. D.; Krasniqi, F.; Bott, M.; Schmidt, K. E.; Wang, X.; Grotjohann, I.; Holton, J. M.; Barends, T. R. M.; Neutze, R.; Marchesini, S.; Fromme, R.; Schorb, S.; Rupp, D.; Adolph, M.; Gorkhover, T.; Andersson, I.; Hirsemann, H.; Potdevin, G.; Graafsma, H.; Nilsson, B. and Spence, J. C. H. (2011). "Femtosecond X-ray protein nanocrystallography". Nature, 470, pp. 73-77. http://dx.doi.org/10.1038/nature09750 |
| ○ | Clemons, W. M. Jr.; Brodersen, D. E.; McCutcheon, J. P.; May, J. L.; Carter, A. P.; Morgan-Warren, R. J.; Wimberly, B. T. and Ramakrishnan, V. (2001). "Crystal structure of the 30 S ribosomal subunit from Thermus thermophilus: purification, crystallization and structure determination". Journal of Molecular Biology, 310, pp. 827-843. http://dx.doi.org/10.1006/jmbi.2001.4778 |
| ○ | Cramer, P.; Bushnell, D. A.; Fu, J.; Gnatt, A. L.; Maier-Davis, B.; Thompson, N. E.; Burgess, R. R.; Edwards, A. M.; David, P. R. and Kornberg, R. D. (2000). "Architecture of RNA polymerase II and implications for the transcription mechanism". Science, 288, pp. 640-649. http://dx.doi.org/10.1126/science.288.5466.640 |
| ○ | Edwards, A. M.; Darst, S. A.; Feaver, W. J.; Thompson, N. E.; Burgess, R. R. and Kornberg, R. D. (1990). "Purification and lipid-layer crystallization of yeast RNA polymerase II". Proceedings of the National Academy of Sciences of the USA, 87, pp. 2122-2126. http://dx.doi.org/10.1073/pnas.87.6.2122 |
| ○ | Franklin, R. E. and Gosling, R. G. (1953). "Molecular Configuration of Sodium Thymonucleate". Nature, 171, pp. 740-741. http://dx.doi.org/10.1038/171740a0 |
| ○ | Goeddel, D. V.; Kleid, D. G.; Bolivar, F.; Heyneker, H. L.; Yansura, D. G.; Crea, R.; Hirose, T.; Kraszewski, A.; Itakura, K. and Riggs, A. D. (1979). "Expression in Escherichia coli of chemically synthesized genes for human insulin". Proceedings of the National Academy of Sciences of the USA, 76, pp. 106-110. http://dx.doi.org/10.1073/pnas.76.1.106 |
| ○ | Grutter, M. G.; Marki, W. and Walliser, H. P. (1985). "Crystals of the complex between recombinant N-acetyleglin c and subtilisin". Journal of Biological Chemistry, 260, pp. 11436-11437. |
| ○ | Itakura, K.; Hirose, T.; Crea, R.; Riggs, A. D.; Heyneker, H. L.; Bolivar, F. and Boyer, H. W. (1977). "Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin". Science, 198, pp. 1056-1063. http://dx.doi.org/10.1126/science.412251 |
| ○ | Johansson, L. C.; Arnlund, D.; White, T. A.; Katona, G.; DePonte, D. P.; Weierstall, U.; Doak, R. B.; Shoeman, R. L.; Lomb, L.; Malmerberg, E.; Davidsson, J.; Nass, K.; Liang, M.; Andreasson, J.; Aquila, A.; Bajt, S.; Barthelmess, M.; Barty, A.; Bogan, M. J.; Bostedt, C.; Bozek, J. D.; Caleman, C.; Coffee, R.; Coppola, N.; Ekeberg, T.; Epp, S. W.; Erk, B.; Fleckenstein, H.; Foucar, L.; Graafsma, H.; Gumprecht, L.; Hajdu, J.; Hampton, C. Y.; Hartmann, R.; Hartmann, A.; Hauser, G.; Hirsemann, H.; Holl, P.; Hunter, M. S.; Kassemeyer, S.; Kimmel, N.; Kirian, R. A.; Maia, F. R. N. C.; Marchesini, S.; Martin, A. V.; Reich, C.; Rolles, D.; Rudek, B.; Rudenko, A.; Schlichting, I.; Schulz, J.; Seibert, M. M.; Sierra, R. G.; Soltau, H.; Starodub, D.; Stellato, F.; Stern, S.; Strüder, L.; Timneanu, N.; Ullrich, J.; Wahlgren, W. Y.; Wang, X.; Weidenspointner, G.; Wunderer, C.; Fromme, P.; Chapman, H. N.; Spence, J. C. H. and Neutze, R. (2012). "Lipidic phase membrane protein serial femtosecond crystallography". Nature Methods, 9, pp. 263-265. http://dx.doi.org/10.1038/nmeth.1867 |
| ○ | Jordan, P.; Fromme, P.; Witt, H. T.; Klukas, O.; Saenger, W. and Krauss, N. (2001). "Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution". Nature, 411, pp. 909-917. http://dx.doi.org/10.1038/35082000 |
| ○ | Kendrew, J. C.; Bodo, G.; Dintzis, H. M.; Parrish, R. G.; Wyckoff, H. and Phillips, D. C. (1958). "A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis". Nature, 181, pp. 662-666. http://dx.doi.org/10.1038/181662a0 |
| ○ | Laue, Max von (1913). "Kritische Bemerkungen zu den Deutungen der Photogramme von Friedrich und Knipping". Physikalische Zeitschrift, 14, pp. 421-423. |
| ○ | Lesley, S. A.; Kuhn, P.; Godzik, A.; Deacon, A. M.; Mathews, I.; Kreusch, A.; Spraggon, G.; Klock, H. E.; McMullan, D.; Shin, T.; Vincent, J.; Robb, A.; Brinen, L. S.; Miller, M. D.; McPhillips, T. M.; Miller, M. A.; Scheibe, D.; Canaves, J. M.; Guda, C.; Jaroszewski, L.; Selby, T. L.; Elsliger, M. A.; Wooley, J.; Taylor, S. S.; Hodgson, K. O.; Wilson, I. A.; Schultz, P. G. and Stevens, R. C. (2002). "Structural genomics of the Thermotoga maritime proteome implemented in a high-throughput structure determination pipeline". Proceedings of the National Academy of Sciences of the USA, 99, pp. 11664-11669. http://dx.doi.org/10.1073/pnas.142413399 |
| ○ | Maier, T.; Jenni, S. and Ban, N. (2006). "Architecture of mammalian fatty acid synthase at 4.5 Å resolution". Science, 311, pp. 1258-1262. http://dx.doi.org/10.1126/science.1123248 |
| ○ | Makde, R. D.; England, J. R.; Yennawar, H. P. and Tan, S. (2010). "Structure of RCC1 chromatin factor bound to the nucleosome core particle". Nature, 467, pp. 562-566. http://dx.doi.org/10.1038/nature09321 |
| ○ | Matsuda, S.; Kawano, G.; Itoh, S.; Mitsui, Y. and Iitaka, Y. (1986). "Crystallization and preliminary X-ray studies of recombinant murine interferon-β". Journal of Biological Chemistry, 261, pp. 16207-16209. |
| ○ | McMullan, G.; Chen, S.; Henderson, R. and Faruqi, A. R. (2009). "Detective quantum efficiency of electron area detectors in electron microscopy". Ultramicroscopy, 109, pp. 1126-1143. http://dx.doi.org/10.1016/j.ultramic.2009.04.002 |
| ○ | Miller, D. L.; Kung, H. F.; Staehelin, T. and Pestka, S. (1981). "The crystallization of recombinant human leukocyte interferon A". Methods Enzymology, 79, pp. 3-7. http://dx.doi.org/10.1016/S0076-6879(81)79005-0 |
| ○ | Miller, D. L.; Kung, H. F. and Pestka, S. (1982). "Crystallization of recombinant human leukocyte interferon A". Science, 215, pp. 689-690. http://dx.doi.org/10.1126/science.6173922 |
| ○ | Morrow, J. F.; Cohen, S. N.; Chang, A. C.; Boyer, H. W.; Goodman, H. M. and Helling, R. B. (1974). "Replication and transcription of eukaryotic DNA in Escherichia coli". Proceedings of the National Academy of Sciences of the USA, 71, pp. 1743-1747. http://dx.doi.org/10.1073/pnas.71.5.1743 |
| ○ | Mueller, M.; Wang, M. and Schulze-Briese, C. (2012). "Optimal fine Φ-slicing for single-photon-counting pixel detectors". Acta Crystallographica D Biological Crystallography, 68, pp. 42-56. http://dx.doi.org/10.1107/S0907444911049833 |
| ○ | Mullis, K. B.; Erlich, H. A.; Arnheim, N.; Horn, G. T.; Saiki, R. K. and Scharf, S. J. (1987). "Process for amplifying, detecting, and/or-cloning nucleic acid sequences". U.S. Patent 4,683,195. |
| ○ | Muñoz, I. G.; Yébenes, H.; Zhou, M.; Mesa, P.; Serna, M.; Park, A. Y.; Bragado-Nilsson, E.; Beloso, A.; de Cárcer, G.; Malumbres, M.; Robinson, C. V.; Valpuesta, J. M. and Montoya, G. (2011). "Crystal structure of the open conformation of the mammalian chaperonin CCT in complex with tubulin". Nature Structural & Molecular Biology, 18, pp. 14-19. http://dx.doi.org/10.1038/nsmb.1971 |
| ○ | Perutz, M. F.; Rossmann, M. G.; Cullis, A. F.; Muirhead, H.; Will, G. and North, A. C. T. (1960). "Structure of Hæmoglobin: A Three-Dimensional Fourier Synthesis at 5.5-Å. Resolution, Obtained by X-Ray Analysis". Nature, 185, pp. 416-422. http://dx.doi.org/10.1038/185416a0 |
| ○ | Pomeranz-Krummel, D. A.; Oubridge, C.; Leung, A. K.; Li, J. and Nagai, K. (2009). "Crystal structure of human spliceosomal U1 snRNP at 5.5 Å resolution". Nature, 458, pp. 475-480. http://dx.doi.org/10.1038/nature07851 |
| ○ | Roberts, R. J. (2005). "How restriction enzymes became the workhorses of molecular biology". Proceedings of the National Academy of Sciences of the USA, 102, pp. 5905-5908. http://dx.doi.org/10.1073/pnas.0500923102 |
| ○ | Sali, A. and Kuriyan, J. (1999). "Challenges at the frontiers of structural biology". Trends in Cell Biology, 9, pp. M20-M24. http://dx.doi.org/10.1016/S0962-8924(99)01685-2 |
| ○ | Sibanda, B. L.; Chirgadze, D. Y. and Blundell, T. L. (2010). "Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats". Nature, 463, pp. 118-121. http://dx.doi.org/10.1038/nature08648 |
| ○ | Śledź, P.; Unverdorben, P.; Beck, F.; Pfeifer, G.; Schweitzer, A.; Förster, F. and Baumeister, W. (2013). "Structure of the 26S proteasome with ATP-γS bound provides insights into the mechanism of nucleotide-dependent substrate translocation". Proceedings of the National Academy of Sciences of the USA, 110, pp. 7264-7269. http://dx.doi.org/10.1073/pnas.1305782110 |
| ○ | Watson, J. D. and Crick, F. H. (1953a). "Molecular structure of nucleic acids, a structure for deoxyribose nucleic acid". Nature, 171, pp. 737-738. http://dx.doi.org/10.1038/171737a0 |
| ○ | Watson, J. D. and Crick, F. H. (1953b). "Genetical implications of the structure of deoxyribonucleic acid". Nature, 171, pp. 964-967. http://dx.doi.org/10.1038/171964b0 |
| ○ | Wilkins, M. H. F.; Stokes, A. R. and Wilson, H. R. (1953). "Molecular Structure of Nucleic Acids: Molecular Structure of Deoxypentose Nucleic Acids". Nature, 171, pp. 738-740. http://dx.doi.org/10.1038/171738a0 |
|