CELEBRATING 100 YEARS OF MODERN CRYSTALLOGRAPHY / CIEN AÑOS DE CRISTALOGRAFÍA MODERNA
CRYSTALLOGRAPHIC STRUCTURE SOLUTION
Claudia Millán
Institute of Molecular Biology of Barcelona (IBMB)
Consejo Superior de Investigaciones Científicas
cmncri@ibmb.csic.es
Isabel Usón
Institute of Molecular Biology of Barcelona (IBMB)
Consejo Superior de Investigaciones Científicas
Institució Catalana de Recerca i Estudis Avançats
uson@ibmb.csic.es
ABSTRACT
The three-dimensional view of molecules at the atomic level provided by X-ray crystallography is not only extremely informative but is also easily and intuitively understood by humans, who very much rely on their vision. However, unlike microscopy, this technique does not directly yield an image. The structural model cannot be directly calculated from the diffraction data, as only the intensities of scattered beams and not their phases are experimentally accessible. In order to obtain the 3-dimensional structure phases have to be obtained by either additional experimental or computational methods. This is known as the phase problem in crystallography. In this manuscript we provide an overview of major milestones along the quest for the lost phases.
RESOLUCIÓN DE ESTRUCTURAS CRISTALOGRÁFICAS
RESUMEN
La cristalografía proporciona una visión tridimensional de las moléculas a un nivel de detalle atómico, que no sólo resulta muy informativa sino que además puede ser fácil e intuitivamente comprendida por seres tan predominantemente visuales como solemos ser los humanos. Sin embargo, al contrario que la microscopía, esta técnica no ofrece directamente una imagen y el modelo estructural no puede calcularse directamente a partir de los datos de difracción, ya que solamente las intensidades de los rayos difractados y no sus fases son accesibles a la medida experimental. Para determinar la estructura tridimensional las fases deben ser obtenidas por medio de métodos adicionales, bien experimentales o computacionales. Esto constituye el problema de la fase en cristalografía. En este artículo ofreceremos una visión general de los principales hitos en la búsqueda de las fases perdidas.
Received: September 12, 2014; Accepted: February 13, 2015.
Citation/Cómo citar este artículo: Millán, C.; Usón, I. (2015). "Crystallographic Structure Solution". Arbor, 191 (772): a218. doi: http://dx.doi.org/10.3989/arbor.2015.772n2004
KEYWORDS: phase problem; constraints; structure factors; Fourier maps; search; minimization; maximum-likelihood; optimization; restraints; X-rays.
PALABRAS CLAVE: problema de la fase; constricciones; factores de estructura; mapas de Fourier; métodos de búsqueda; minimización; máxima verosimilitud; optimización; restricciones; rayos-X.
Copyright: © 2015 CSIC. This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial (by-nc) Spain 3.0 License.
CONTENTS
INTRODUCTION
Top
In 1914 Max von Laue received the Nobel Prize in Physics “for his discovery of the diffraction of X-rays by crystals” (Friedrich, Knipping and Laue, 1912Friedrich, W.; Knipping, P. and Laue, M. (1912). "Interferenz-Erscheinungen bei Röntgenstrahlen". Sitzungsberichte der Königlich Bayerische Akademie der Wissenschaften, pp. 303-322.; von Laue, 1912von Laue, M. (1912). "Eine quantitative prüfung der theorie für die interferenz-erscheinungen bei Röntgenstrahlen". Sitzungsberichte der Königlich Bayerische Akademie der Wissenschaften, pp. 363-373.). This discovery marked the beginning of X-ray crystallography, which to this day has remained an essential tool of investigation throughout the sciences because it provides conclusive information on molecular structure down to the atomic level. From its onset, 100 years ago (Bragg and Bragg, 1913aBragg, W. H. and Bragg, W. L. (1913a). "The Reflexion of X-rays by Crystals". Proceedings of the Royal Society of London A, 88 (605), pp. 428-438, http://dx.doi.org/10.1098/rspa.1913.0040.), crystallography has transformed our understanding of the natural sciences and has become indispensable by providing a view into the three-dimensional molecular structures that is yet unsurpassed by any other structural technique in its degree of detail and precision. Furthermore, crystallography uniquely complements structural information from other methods suiting different conditions, such as nuclear magnetic resonance in solution and solid-state studies, small angle scattering in solution or low-resolution electron microscopy from complex systems. For such a species as us, humans, for whom in most cases vision constitutes the predominant sense in our apprehension of the physical world, it is evident how the visualization of the main players in the chemical reaction processes inherent to nature and life, provides a powerful frame to relate all our functional knowledge and understand underlying mechanisms. Nevertheless, contrarily to what happens for instance in microscopy, the product of the crystallographic analysis is not a direct image of the molecules in the crystal. In the diffraction experiment, only the scattered intensities and not the phases from the X-rays, neutrons or electrons are directly measurable. However, the phases for each diffracted beam are essential to structure determination: without them the three-dimensional structure cannot be computed. This gives rise to the phase problem, central to crystallography. Obtaining the missing phases has ever been a quest in the crystallographic forefront. Even though in the last century a number of ways to solve the phase problem have been developed, the large number of parameters in today’s complex problems and the frequent limitations in the data quality attainable in challenging studies still tend to hamper structure solution and phasing becomes a bottleneck in structure determination. In this manuscript we present an overview of the quest for the lost phases leading to structure solution.
HOW ESSENTIAL ARE PHASES AND WHY PHASING IS DIFFICULT
Top
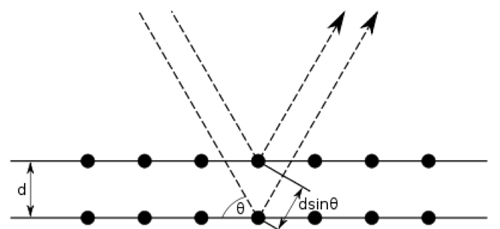
Given a crystal of known structure, it was soon realized how to calculate the effect of diffraction of X-rays by the electrons present (Bragg, 1913aBragg, W. L. (1913a). "The Diffraction of Short Electromagnetic Waves by a Crystal". Mathematical Proceedings of the Cambridge Philosophical Society, 17, pp. 43-57.; Ewald, 1913Ewald, P. P. (1913). "About the theory of the interference of X-rays in crystals (Zur Theorie der interferenzen der Röntgen-strahlen in kristallen)". Physikalische Zeitschrift, 14, pp. 465-472.) (or the scattering of neutrons by the atomic nuclei). Bragg developed an intuitive and simple mathematical description of the diffraction of X-rays by crystals in terms of reflection by lattice planes separated by a spacing d, at a glancing angle θ as the condition for constructive interference, hence deriving what is now known as Bragg’s law: nλ = 2d sin θ (Figure 1). The minimal spacing between lattice planes for which diffraction data can be recorded for a given crystal is called maximal resolution or high resolution limit, and it approximately matches the resolvability of structural features in the resulting electron density map.
|
Figure 1. Bragg diffraction. Two beams with identical wavelength and phase approach a crystalline solid and are scattered by two lattice planes. The lower beam traverses an extra length of 2dsinθ. When this length is equal to an integer multiple of the wavelength of the radiation, constructive interference occurs.
[Descargar tamaño completo]
| |
The resolution limit is related to the experimental setup but more essentially to the crystal properties, imperfect ordering bringing about a loss of resolution. The amount of unique experimental data that can be recorded from a crystal is limited by the maximal resolution to which it diffracts: the higher the resolution (the smaller d), the more independent data are measurable and thus, the more parameters can be afforded to characterize the structural model.
The fundamental relationship between experimental data and the electron density function in the crystal is given by the Fourier transformation of the individual structure factors F. Each structure factor F is a complex number with amplitude and phase; however, since the measured intensities are proportional to the squared structure factors the phase information is lost. The intensities of the scattered beams recorded would be roughly proportional to the square of the structure factors (subject to predictable corrections to account for experimental conditions). But the objective of a crystallographic determination is to resolve the inverse problem of determining the molecular structure within the crystal from the intensities of the scattered beams. To compute the corresponding inverse Fourier transform, with the structure factors as coefficients, their phases should be known as well as their moduli.
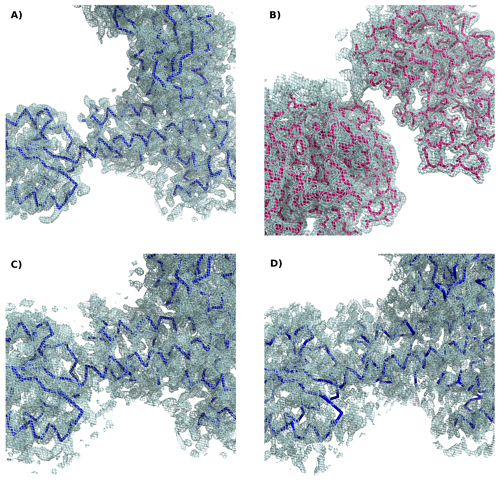
To understand the importance of the phase information content, it is very instructive to calculate a Fourier transform with amplitudes corresponding to one structure and phases derived from a different one. Figure 2 illustrates this in the case of two molecules.
|
Figure 2. Electron density maps calculated for: a) Experimental amplitudes for the protein TsaR with final phases from TsaR. B) Amplitudes for the protein Windbeutel with phases from Windbeutel set in a TsaR-like crystal. C) Amplitudes from Windbeutel with phases from TsaR: the structure of TsaR but not that of Windbeutel is recognizable. D) 3% most intense amplitudes from all resolution shells of TsaR and final phases from TsaR: a map calculated with the most intense data and correct phases provides a good approximation.
[Descargar tamaño completo]
| |
For a detailed pictorial explanation of Fourier transforms, phases and amplitudes in diffraction see the excellent Kevin Cowtan’s Picture Book of Fourier Transforms (http://bit.ly/1KDJNqz).
It is important to realize that as a consequence of the Fourier transformation each atom contributes to all structure factors, each structure factors carries information about all atoms. This confers structure solution an “all or nothing” quality: even though phasing does not usually provide the final model as interpretation of the electron density map -refinement and validation are still required- it affords an overall, fairly complete view characterizing a substantial part of the structure. In other words, all strong data need to be accounted for simultaneously, all prominent features in the electron density need to be determined simultaneously as one portion of the structure is not exclusively related to a subset of data or vice versa.
In addition to the phase problem inherent to diffraction, experimental data may be of limited quality (finite resolution, incomplete data, crystal pathologies such as twinning, disorder, pseudo-symmetry or anisotropic diffraction, errors in measured intensities), which in turn may complicate phasing.
SO HOW WERE THE FIRST STRUCTURES SOLVED?
Top
If phases cannot be measured but are needed to calculate the electron density map from which the structural model is interpreted, how have we come to determine and archive in repositories over 173.000 structures of minerals (http://bit.ly/1DAgMdk), 750.000 structures of small organic and organometallic compounds (Allen, 2002Allen, F. H. (2002). "The Cambridge Structural Database: a quarter of a million crystal structures and rising". Acta Crystallographica, B58, pp. 380-388, http://dx.doi.org/10.1107/S0108768102003890.) (http://bit.ly/1E27p3Y) and 100.000 structures of proteins and nucleic acids, that is macromolecules (Bernstein et al. 1977Bernstein, F. C.; Koetzle, T. F.; Williams, G. J. B.; Meyer, E. F.; Brice, M. D.; Rodgers, J. R.; Kennard, O.; Shimanouchi, T. and Tasumi, M. (1977). "The Protein Data Bank: a computer-based archival file for macromolecular structures". Journal of Molecular Biology, 112, pp. 535-542, http://dx.doi.org/10.1016/S0022-2836(77)80200-3.; Berman et al., 2000Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N. and Bourne, P. E. (2000). "The Protein Data Bank". Nucleic Acids Research, 28, pp. 235-242, http://dx.doi.org/10.1093/nar/28.1.235.) (http://bit.ly/1vg7iNm).
Given a set of experimentally derived amplitudes (=Structure factors), we can postulate that there is only one chemically plausible structure in the crystal that is consistent with these experimental data (Giacovazzo, 2011Giacovazzo, C. (2011). Fundamentals of crystallography (3rd ed). Oxford, New York: Oxford University Press - International Union of Crystallography texts on crystallography, http://dx.doi.org/10.1093/acprof:oso/9780199573653.001.0001.). If that were not the case, we could not have certainty that our structural analysis would be conclusive, but so far, no such two different structures giving rise to the same diffraction have been found. Therefore, and as we are able to calculate the effect of diffraction on a crystal, if we could setup a model of the structure in the crystal, we could verify its correctness. This is precisely how the first structures were solved: by realizing the only possible way in which periodic structures composed by one or two different atoms could be arranged in order to cause the symmetry and intensity of a particular diffraction pattern. William Henry Bragg and William Lawrence Bragg, father and son, published in 1913 the structures of the first inorganic compounds, those of diamond (Bragg and Bragg, 1913bBragg, W. H. and Bragg, W. L. (1913b). "The structure of the diamond". Nature, 91, pp. 557, http://dx.doi.org/10.1038/091557a0.), and those of sodium chloride, potassium chloride, and potassium bromide (W. L. Bragg, 1913bBragg, W. H. and Bragg, W. L. (1913b). "The structure of the diamond". Nature, 91, pp. 557, http://dx.doi.org/10.1038/091557a0.). Structures where not all the atomic positions were constrained by the symmetry, such as iron pyrite FeS2, were adjusted by trial and error (Bragg, 1925Bragg, W. L. (1925). "The Crystalline Structure of Inorganic Salts". Nature, 116, pp. 557, http://dx.doi.org/10.1038/116249a0.). It is remarkable that, even though these reticular structures without discrete molecules were at the time, perceived by chemists as strongly contradicting their molecular theory, they were swiftly accepted albeit not without some opposition (Armstrong, 1927Armstrong, H. E. (1927). "Poor Common Salt!". Nature, 120, pp. 478, http://dx.doi.org/10.1038/120478b0.). As humans, we have learned to understand by seeing.
Many years later, in the early 50s the same principle of combined atomic understanding, learned from crystal structures of peptide building blocks, with constraints from fiber diffraction (Astbury and Street, 1932Astbury, W. T. and Street, A. (1932). "A. X-Ray studies of the structure of hair, wool, and related fibers. I. General". Philosophical Transactions of the Royal Society A, 230, pp. 75-101, http://dx.doi.org/10.1098/rsta.1932.0003.), led Pauling to propose the structures of the α-helix (Pauling, Corey and Branson, 1951Pauling, L.; Corey, R. B. and Branson, H. R. (1951). "The structure of proteins, two hydrogen-bonded helical configurations of the polypeptide chain". Proceedings of the National Academy of Sciences of the USA, 37, pp. 205-211, http://dx.doi.org/10.1073/pnas.37.4.205.) and β-sheets in polypeptides in proteins (Pauling and Corey, 1951Pauling, L. and Corey, R. B. (1951). "The pleated sheet, a new layer configuration of polypeptide chains". Proceedings of the National Academy of Sciences of the USA, 37, pp. 251-256, http://dx.doi.org/10.1073/pnas.37.5.251.). This diffraction-constrained model building culminated with the first atomic model for a much more complicated structure. The DNA double helix was proposed based on the analysis of fiber diffraction patterns (Franklin and Gosling, 1953Franklin, R. and Gosling, R. G. (1953). "Molecular Configuration in Sodium Thymonucleate". Nature, 171, pp. 740-741, http://dx.doi.org/10.1038/171740a0.) and consideration that only a right-handed, base-paired double helix would be able to produce it (Watson and Crick, 1953Watson, J. D. and Crick, F. H. C. (1953). "Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid". Nature, 171, pp. 737-738, http://dx.doi.org/10.1038/171737a0.). Curiously enough, the first atomic structure of a DNA fragment determined by X-ray crystallography, that of Z-DNA turned out to be left handed (Wang et al., 1979Wang, A. H.-J.; Quigley, G. J.; Kolpak, F. J.; Crawford, J. L.; van Boom, J. H.; van der Marel, G. and Rich, A. (1979). "Molecular structure of a left-handed double helical DNA fragment at atomic resolution". Nature, 282, pp. 680-686, http://dx.doi.org/10.1038/282680a0.).
Beyond such cases, where symmetry and boundary conditions led to establishing a model susceptible of being validated by the data, most phasing problems had to start from the Fourier analysis of the experimental intensity data.
WHAT CAN BE CALCULATED FROM THE DATA: THE PATTERSON FUNCTION
Top
The experimental data allow direct computing of a function that has immediate physical meaning. A Fourier transform calculated using as coefficients the square values of the structure factors, that is, quantities that are proportional to the recorded intensities is thus phase independent (Patterson, 1935Patterson, A. L. (1935). "A direct method for the determination of the components of interatomic distances in crystals". Zeitschrift für Kristallographie, 90, pp. 517-542, http://dx.doi.org/10.1524/zkri.1935.90.1.517.). The physical meaning of the Patterson function corresponds to superimposing two copies of the electron density in the unit cell, shifted by a variable translation. Its maxima will result from all possible interatomic vectors, translated to an arbitrary origin. The Patterson function will have a trivial maximum for a translation value of 0 in all directions, as evidently this corresponds to perfectly superimposing two copies of the electron density, in which case every atom will be correlated to itself. For non-zero translations, the value of the Patterson function will be markedly higher if it leads to superimposition of heavy atoms with many electrons, as it is proportional to the product of the atomic numbers of correlated atoms or –accidental or systematic– superposition of parallel interatomic vectors of the same length. The Patterson function opened the door to the determination of structures containing one or few markedly heavier atoms within a small structure, as their positions could be directly calculated, taking symmetry into account (Harker, 1936Harker, D. (1936). "The application of the three-dimensional Patterson method and the crystal structures of proustite, Ag3AsS3, and pyrargyrite, Ag3SbS3". The Journal of Chemical Physics, 4, pp. 381-390, http://dx.doi.org/10.1098/rspa.1954.0203.). From these initial atomic sites, approximate phases could be derived and in addition to the heavy atoms, remaining atoms could be found in the electron density maps calculated with the measured amplitudes and heavy atom phases. Iterating the process as more of the structure could be interpreted allowed, with this “heavy atom method” to complete the structure.
Tagging the molecule with a heavy atom having so many electrons that it would dominate scattering provided a way to phasing chemical structures. Still, relatively small equal atom structures remained difficult until the advent of direct methods.
Dorothy Hodgkin’s determination of the structure of penicillin through the sodium and rubidium salts of benzylpenicillin (Crowfoot et al., 1949Crowfoot, D.; Bunn, C. W.; Rogers-Low, B. W. and Turner-Jones, A. (1949). "X-ray crystallographic investigation of the structure of penicillin". In Clarke, H. T.; Johnson, J. R. and Robinson, R. (eds.), Chemistry of Penicillin, pp. 310-367. Princeton University Press.) was both a major challenge and a chemical puzzle as well as a priority for its biomedical impact. With 41 independent atoms, penicillin was the largest molecule to have been determined by X-ray diffraction at the time it was solved. Hodgkin received the Nobel Prize in 1964, having also elucidated the structure of vitamin B12 (Hodgkin et al., 1956Hodgkin, D. C.; Kamper, J.; Mackay, M.; Pickworth, J.; Trueblood, K. N. and White, J. G. (1956). "Structure of vitamin B-12". Nature, 178, pp. 64-66, http://dx.doi.org/10.1038/178064a0.), among other biological molecules and later on insulin (Adams et al., 1969Adams, M. J.; Blundell, T. L.; Dodson, E. J.; Dodson, G. G.; Vijayan, M.; Baker, E. N.; Harding, M. M.; Hodgkin, D. C.; Rimmer, B. and Sheat, S. (1969). "Structure of Rhombohedral 2 Zinc Insulin Crystals". Nature, 224, pp. 491, http://dx.doi.org/10.1038/224491a0.).
DIRECT METHODS
Top
In the case of well diffracting crystals of small molecules, the number of independent diffraction data that can be measured is much higher than the number of parameters required to describe the positions of all atoms in the molecule to be determined. The fact that the system is overdetermined implies that a solution may be possible if the experimentally accessible structure factors are related by a set of known relationships.
Overdetermination can be exploited as the possible sets of phases are not independent. Conditions required for a solution to have physical meaning can be applied as constraints on the sets of phases. Applying these conditions such as positivity (in every point, the electron density must have a non-negative value: either there are some electrons or there are none) and atomicity (structures are composed of atoms) leads to certain statistical relationships among phases.
The first mathematical conditions to be recognized were the inequalities by Harker and Kasper (1948Harker, D. and Kasper, J. S. (1948). "Phases of Fourier coefficients directly from crystal diffraction data". Acta Crystallographica, 1, pp. 70-75, http://dx.doi.org/10.1107/S0365110X4800020X.), followed by the positivity derived determinants by Karle and Hauptman (1950Karle, J. and Hauptman, H. (1950). "The phases and magnitudes of the structure factors". Acta Crystallographica, 3, pp. 181-187, http://dx.doi.org/10.1107/S0365110X50000446.) but the turning point came with Sayre’s equation (Sayre, 1952Sayre, D. (1952). "The squaring method: a new method for phase determination". Acta Crystallographica, 5, pp. 60-65, http://dx.doi.org/10.1107/S0365110X52000137.), which is based on the assumption of a structure formed by equal, point-like atoms. For such a structure, its square electron density function would be proportional to the electron density function. The latter adopting in each point either the value 0 or that of the electrons Z in the atom, its square must necessarily be either 02 or Z2, which is the same as the constant Z times 0 or Z. From this expression, the triplet formula relating the phases of three strong reflections whose indices h, k and h-k (coordinates in reciprocal space) add up to zero, ϕ(h) ≈ ϕ(k)+ ϕ(h-k) can be derived. Phase relationships and their probabilities are combined in the tangent formula, the most efficient phase search motor in direct methods (Hauptman and Karle, 1953Hauptman, H. and Karle, J. (1953). Solution of the phase problem I. The centrosymmetric crystal. Dayton, Ohio: American Crystallographic Association.; Karle and Hauptman, 1956Karle, J. and Hauptman, H. (1956). "A theory of phase determination for the four types of non-centrosymmetric space groups 1P222, 2P22, 3P12, 3P22". Acta Crystallographica, 9, pp. 635-651, http://dx.doi.org/10.1107/S0365110X56001741.).
In practice, a good approximation to the function describing the electron density can be calculated from roughly the 3% most intense reflections in every resolution shell (Figure 2). Multisolution methods are based on the random assignment of phase values to a subset of strong reflections, followed by refinement of these values to best fulfill the atomicity constraint and extend phases to all unique reflections interpreting and completing the solution in terms of atoms. This procedure is repeated until a mathematical and chemically reasonable solution is found. Herbert Hauptman and Jerome Karle were awarded the Nobel Prize in 1984 for their derivation of the fundamental equations underlying today’s direct methods for phasing crystal structures.
Computer implementations of direct methods, such as SHELXS (Sheldrick, 2008Sheldrick, G. M. (2008). "A short history of SHEL". Acta Crystallographica, A64, pp. 112-122, http://dx.doi.org/10.1107/S0108767307043930.) dominate phasing in small molecule crystallography. Recently, charge flipping algorithms have been introduced, which, also easily solve equal atom structures up to 250 atoms. Charge flipping is an iterative Fourier cycle that unconditionally modifies the calculated electron density and structure factors in real and reciprocal space, and even if less efficient than direct methods, it has become feasible through the increase in computing power (Oszlányi and Süto, 2004Oszlanyi, G. and Süto, A. (2004). "Ab initio structure solution by charge flipping". Acta Crystallographica, A60, pp. 134-141, http://dx.doi.org/10.1107/S0108767303027569.). In the field of chemical crystallography, where the structure to be solved usually contains less than 200 independent atoms and crystals almost invariably diffract to atomic resolution, these days most cases are automatically solved by direct methods or charge flipping in fractions of a second, yielding a practically complete model.
MACROMOLECULES, WHY ARE THEY DIFFICULT TO SOLVE AND WHAT TO DO
Top
In the field of biological crystallography, where the molecules crystallized are proteins, nucleic acids and their complexes, the situation is very different. Although the first diffraction images from a hydrated protein crystal were recorded as early as 1934 (Bernal and Crowfoot, 1934Bernal, J. D. and Crowfoot, D. (1934). "X-Ray Photographs of Crystalline Pepsin". Nature, 133, pp. 794-795, http://dx.doi.org/10.1038/133794b0.), it would take more than 20 years before the first atomic structure of a protein could be determined. The macromolecular structures to be determined are more complex, requiring a larger number of atoms to be simultaneously found. Even a comparably small protein, such as lysozyme, contains 1000 independent atoms and, frequently, several copies of far larger structures are found in the asymmetric unit. Also, macromolecular crystals are grown in aqueous solutions and typically half of the crystal volume is occupied by water disrupting internal order and hence diffraction properties. Protein crystals cannot be dried, as this will destroy the crystal and they are in general mechanically fragile and more delicate than mineral or chemical crystals. As this aqueous solution is largely disordered and does not share the periodicity of the crystal, it does not contribute to the intensities of the diffracted beams in the way ordered atoms do. Yet, it is not the same as if the space not occupied by the molecule was void and solvent scattering further complicates diffraction. The exposure of the macromolecular surface to this solution allows motion of mobile parts, such as solvated side chains or loops and induces differences among crystallographically equivalent molecules. The periodicity in the crystal being less perfect, differences start to dominate as the crystal is examined in finer detail. This is reflected in the ability to diffract: on one hand, weak reflections become drowned in the background scattering derived from diffuse solvent and crystal mount and on the other, diffraction breaks up at a given resolution. Indeed, it is but a small fraction of macromolecules, fewer than 2.5% of the structures deposited with the PDB that diffracts to 1.2 Å, considered to be the limit of atomic resolution. In addition, this water solution favors the propagation of radiation induced radical reactions, which take place during the diffraction experiment. Even though nowadays macromolecular data are generally recorded on crystals frozen to -173º C, radiation damage is still a major problem and has severe consequences on the quality of the diffraction data.
In summary, macromolecules pose a difficult problem as a consequence of the much larger size of the structure, lower data quality and resolution and more fragile crystals. The first approach opening a way into protein structures was a divide and conquer one: inducing a small molecule into the macromolecule and solving the phase problem in two steps.
EXPERIMENTAL PHASING WITH DERIVATIVES: WAYS TO INTRODUCE A SMALL MOLECULE INTO THE MACROMOLECULE
Top
The aqueous solution composing half of the protein crystal volume, whose negative effects have just been described, can also be exploited to mediate the solution to the phase problem. Crystals are maintained in the solution where they were grown to prevent dehydration. Incorporating into this solution a chemical species containing heavy atoms, such as soluble platinum, gold, mercury or uranium salts and complexes may lead to the selective incorporation of such species into particular positions of the macromolecule, so that they become part of the periodic structure and contribute to Bragg diffraction. Diffraction can be recorded on crystals upon such treatment and if local, rather than large scale changes are brought upon, the differences in the structure factors between the native and derivatized crystals can be used as an approximation to the diffraction of the heavy atom substructure. This substructure, if composed by a few ordered, heavy atoms, can be solved from the difference data by small molecule methods.
Once the substructure is solved, phase information can be derived for the native macromolecule through trigonometric relations among the recorded data and the determined heavy atom structure factors. Two independent derivatives (with different substructures) are required in theory to determine the sought phases. David Harker provided the methodology for this kind of phase analysis in 1956 while studying the structure of the protein ribonuclease (Harker, 1956Harker, D. (1956). "The determination of the phases of the structure factors of non-centrosymmetric crystals by the method of double isomorphous replacement". Acta Crystallographica, 9, pp. 1-9, http://dx.doi.org/10.1107/S0365110X56000012.). In practice, many more derivatives may be needed as in real crystals errors may dominate, obscuring the phase information. Often, the soaking process may damage the crystals and spoil their diffraction properties, fail to incorporate ordered heavy atoms or induce structural changes precluding its combination with the native data, to name just some of the most frequent hurdles. On the other hand, occasionally a single derivative has been used to successfully phase the native structure (SIR).
This method, named Multiple Isomorphous Replacement (MIR) was used to determine the first protein structures at the MRC in Cambridge, those of Myoglobin and Hemoglobin by Kendrew and Perutz (Green, Ingram and Perutz, 1954Green, D. W.; Ingram, V. M. and Perutz, M. F. (1954). "Structure of haemoglobin: IV. Sign determination by the isomorphous replacement method". Proceedings of the Royal Society of London. Series A, 225, pp. 287-307, http://dx.doi.org/10.1098/rspa.1954.0203.; Kendrew et al., 1958Kendrew, J. C.; Bodo, G.; Dintzis, H. M.; Parrish, R. G.; Wyckoff, H. and Phillips, D. C. (1958). "A three dimensional model of the myoglobin molecule obtained by X-ray analysis". Nature, 181, pp. 662-666, http://dx.doi.org/10.1038/181662a0.).
To overcome the difficulties in extracting a weak signal from the necessarily noisy difference intensities recorded from native and soaked crystals, the alternative is recording diffraction data on crystals of the same kind or even on the same specimen but under conditions that will modify the diffraction properties of particular elements present in the sample. By choosing appropriate wavelengths from a tunable X-ray source, dispersive and anomalous scattering contributions of particular elements may be amplified enough to modify the recorded intensities by a small percentage. In this case, rather than inducing a different substructure in various crystals, the diffraction properties of the same substructure are modified in each diffraction experiment and again, the differences brought upon can be exploited to determine the substructure by small-molecule methods and establish relationships to the phases of the macromolecular structure. This method, named Multiple wavelength Anomalous Diffraction (MAD) (Hendrickson, 1991Hendrickson, W. A. (1991). "Determination of Macromolecular Structures from Anomalous Diffraction of Synchrotron Radiation". Science, 254, pp. 51-58, http://dx.doi.org/10.1126/science.1925561.) has gained increasing popularity as tunable beamlines have been popularized and the required precision in measurement of the experimental data has become standard rather than exceptional. Routine data collection on frozen crystals (Hope, 1990Hope, H. (1990). "Crystallography of biological macromolecules at ultra-low temperature". Annual Review of Biophysics and Biophysical Chemistry, 19, pp. 107-126, http://dx.doi.org/10.1146/annurev.bb.19.060190.000543.; Garman and Schneider, 1997Garman, E. F. and Schneider, T. R. (1997). "Macromolecular Cryocrystallography". Journal of Applied Crystallography, 30, pp. 211-237, http://dx.doi.org/10.1107/S0021889897002677.), allowing to collect several datasets on the same specimen before radiation damage has become severe and reliable solutions to incorporate anomalous scatterer substructures have promoted its use. Even though a single heavy atom derivative (SIR) or anomalous scattering recorded at a single wavelength (SAD) should not provide enough data to mathematically determine the sought phases, advances in effective ways to constrain the ambiguous set of starting phases into a physically meaningful solution have allowed SAD to become increasingly popular. These so-called density-modification procedures (Wang, 1985Wang, B.-C. (1985). "Resolution of phase ambiguity in macromolecular crystallography". Methods in Enzymology, 115, pp. 90-112, http://dx.doi.org/10.1016/0076-6879(85)15009-3.; Cowtan and Main, 1993Cowtan, K. D. and Main, P. (1993). "Improvement of macromolecular electron-density maps by the simultaneous application of real and reciprocal space constraints". Acta Crystallographica, D49, pp. 148-157, http://dx.doi.org/10.1107/S0907444992007698.) are used to improve the experimental phases.
Most appropriate elements are again rather electron-rich, from the fourth row onwards in the periodic table. A number of proteins occur containing such elements already in their native form: iron, zinc or molybdenum being the most frequent, but in general the necessity to incorporate a heavy atom substructure would revert to soaking or co-crystallization techniques. Fortunately, substituting methionine by seleno-methionine in recombinant proteins offers a practical solution to eliminate the hurdles brought upon by the rather harsh derivatization treatment on fragile crystals. For chemically synthesized nucleic acids, selective incorporation of bromine in uracil provides an analogous solution.
EXPERIMENTAL PHASING WITH INHERENT ANOMALOUS SIGNAL: NATIVE PHASING
Top
The anomalous scattering of the light carbon, nitrogen and oxygen atoms, which are the main components in proteins, is negligible for phasing purposes. On the contrary, that of the sulfur present in the amino acids cysteine and methionine and that of phosphorous present in nucleic acids is weak but has been shown to be usable since the early 80’s with the determination of the sulfur rich, small protein crambin (Hendrickson and Teeter, 1981Hendrickson, W. A. and Teeter, M. M. (1981). "Structure of the hydrophobic protein crambin determined directly from the anomalous scattering of sulphur". Nature, 290, pp. 107-113, http://dx.doi.org/10.1038/290107a0.). Later on, weak anomalous scatterers present in the aqueous solution of the crystal, such as bromine or iodine were exploited, alone or complementing the anomalous scattering of the sulfur atoms present in the structure (Dauter et al., 1999Dauter, Z.; Dauter, M.; de La Fortelle, E.; Bricogne, G. and Sheldrick, G. M. (1999). "Can anomalous signal of sulfur become a tool for solving protein crystal structures?". Journal of Molecular Biology, 289, pp. 83-92, http://dx.doi.org/10.1006/jmbi.1999.2743.). Methods originally developed for ab initio structure solution (vide infra) were capable of determining substructures composed of a large number of partially occupied and/or weak anomalous scatterers, while sophisticated treatment of errors and phase extension algorithms succeeded in rendering the phases of the native structure from this weak start phase information.
Experimental advances at synchrotron beamlines allowing to collect more precise data through highly redundant datasets (Dauter and Adamiak, 2001Dauter, Z. and Adamiak, D. A. (2001). "Anomalous signal of phosphorus used for phasing DNA oligomer: importance of data redundancy". Acta Crystallographica, D57, pp. 990-995, http://dx.doi.org/10.1107/S0907444901006382.; Broennimann et al., 2006Broenniman, C.; Eikenberry, E. F.; Henrich, B.; Horisberger, G.; Huelsen, G.; Pohl, E.; Schmitt, B.; Schulze-Briese, C.; Suzuki, M.; Tomizaki, T.; Toyokawa, A. and Wagner, A. (2006). "The Pilatus 1M detector". Journal of Synchrotron Radiation, 13, pp. 120-133, http://dx.doi.org/10.1107/S0909049505038665.), maximize the anomalous signal at longer wavelengths (Yang et al., 2003Yang, C.; Pflugrath, J. W.; Courville, D. A.; Stence, C. N. and Ferrara, J. D. (2003). "Away from the edge: SAD phasing from the sulfur anomalous signal measured in-house with chromium radiation". Acta Crystallographica, D59, pp. 1943-1957, http://dx.doi.org/10.1107/S0907444903018547.; Weinert et al., 2015Weinert, T.; Olieric, V.; Waltersperger, S.; Panepucci, E.; Chen, L.; Zhang, H.; Zhou, D.; Rose, J.; Ebihara, A.; Kuramitsu, S.; Li, D.; Howe, N.; Schnapp, G.; Pautsch, A.; Bargsten, K.; Prota, A. E.; Surana, P.; Kottur, J.; Nair, D. T.; Basilico, F.; Cecatiello, V.; Pasqualato, S.; Boland, A.; Weichenrieder, O.; Wang, B.-C.; Steinmetz, M. O.; Caffrey, M. and Wang, M. (2015). "Fast native-SAD phasing for routine macromolecular structure determination". Nature Methods, 12, pp. 131-133, http://dx.doi.org/10.1038/nmeth.3211.), low-noise X-ray detectors (Mueller, Wang and Schulze-Briese, 2012Mueller, M.; Wang, M. and Schulze-Briese, C. (2012). "Optimal fine ϕ-slicing for single-photon-counting pixel detectors". Acta Crystallographica, D68, pp. 42-56, http://dx.doi.org/10.1107/S0907444911049833.) and improvement in the computing programs are pushing a revival of native phasing, this time on much more complex structures than those of crambin or lysozyme. Indeed, structures as large as 1148 independent amino acids have been phased at 2.8 Å from the inherent sulfur in the protein (Liu et al., 2012Liu, Q.; Dahmane, T.; Zhang, Z.; Assur, Z.; Brasch, J.; Shapiro, L.; Mancia, F. and Hendrickson, W. A. (2012). "Structures from Anomalous Diffraction of Native Biological Macromolecules". Science, 336, pp. 1033-1037, http://dx.doi.org/10.1126/science.1218753.). This constitutes a very attractive development, which could become standard given its experimental straightforwardness.
NON CRYSTALLOGRAPHIC SYMMETRY AND MOLECULAR REPLACEMENT
Top
When illustrating the phase problem (Figure 2), the point was made that a map calculated with amplitudes from one structure and phases from another will rather resemble the structure that contributed the phases. This suggests the course of “borrowing” phases from a structure related to the unknown one contained in a crystal. In proteins, the 3-dimensional structure is determined by the amino acid sequence and therefore, if coordinates have been determined for a protein of similar sequence, such as a mutant or an homologous protein from a different organism or one of the components in a complex has been previously characterized, it is possible to exploit this structural knowledge to phase the new target structure.
The phasing problem to be solved is how to place the search model in the unit cell in order to best account for the experimental data recorded (Huber, 1965Huber, R. (1965). "Die automatisierte Faltmolekülmethode". Acta Crystallographica, 19, pp. 353-356, http://dx.doi.org/10.1107/S0365110X65003444.; Rossman, 1972Rossmann, M. G. (1972). The Molecular Replacement Method. New York: Gordon & Breach.). Traditionally, the problem has been divided into the sequential determination of the orientation of the model in the unit cell or rotation search (Rossman and Blow, 1962Rossmann, M. G. and Blow, D. M. (1962). "The Detection of Subunits within the Crystallographic Asymmetric Unit". Acta Crystallographica, 15, pp. 24-31, http://dx.doi.org/10.1107/S0365110X62000067.) and the determination of the translation to be applied to the correctly rotated model or translation search (Crowther and Blow, 1967Crowther, R. A. and Blow, D. W. (1967). "A Method of Positioning a Known Molecule in an Unknown Crystal Structure". Acta Crystallographica, 23, pp. 544-548, http://dx.doi.org/10.1107/S0365110X67003172.). Higher-dimensional searches combining rotation and translation or even simultaneous placement of several fragments are also possible, although computationally much more intensive (Kissinger, Gehlhaar and Fogel, 1999Kissinger, C. L.; Gehlhaar, D. K. and Fogel, D. B. (1999). "Rapid automated molecular replacement by evolutionary search". Acta Crystallographica, D55, pp. 484-491, http://dx.doi.org/10.1107/S0907444998012517.).
The molecular replacement method is intimately related to that of non-crystallographic symmetry. Ultimately, it reduces to exploiting the presence of the same or a similar structure in different crystals or in several crystallographically independent copies present in the same crystal. The structure may be unknown in part or in all of these copies. In the first case, the known stereochemistry is used to construct a model of the unknown crystal. From this, phases can be calculated and map interpretation, model building and refinement will be applied to develop the starting, approximate model into a more accurate representation of the content of the crystal. In the second case, even if the structure is completely unknown, if the relative location of the different copies in one or several polymorphs can be established, it may provide a very powerful constraint on the phases. This is particularly useful to constrain a highly incorrect set of starting phases or in favorable cases, even to drive a random phase set into the correct one. This particular case proved crucial for the structure solution of virus particles (Harrison et al., 1978Harrison, S. C.; Olson, A. J.; Schutt, C. E.; Winkler, F. K. and Bricogne, G. (1978). "Tomato bushy stunt virus at 2.9 Å resolution". Nature, 276, pp. 368-373, http://dx.doi.org/10.1038/276368a0.; Abad- Zapatero et al., 1980Abad-Zapatero, C.; Abdel-Meguid, S. S.; Johnson, J. E.; Leslie, A. G.; Rayment, I.; Rossmann, M. G.; Suck, D. and Tsukihara, T. (1980). "Structure of southern bean mosaic virus at 2.8Å resolution". Nature, 286, pp. 33-39, http://dx.doi.org/10.1038/286033a0.) and viral proteins (Champness et al., 1976Champness, J. N.; Bloomer, A. C.; Bricogne, G.; Butler, P. G. and Klug, A. (1976). "The structure of the protein disk of tobacco mosaic virus to 5A resolution". Nature, 259, pp. 20-24, http://dx.doi.org/10.1038/259020a0.), containing 60 independent copies of the same structural unit organized in a highly ordered, almost spherical geometry, and long anticipated before their atomic structures could be determined.
Molecular replacement has become the main phasing method for macromolecules as the large number of already determined structures increases its applicability. Beyond this obvious advantage, the development of sophisticated and efficient approximations to Maximum likelihood functions (Storoni, McCoy and Read, 2004Storoni, L. C.; McCoy, A. J. and Read, R. J. (2004). "Likelihood-enhanced fast rotation functions". Acta Crystallographica, D60, pp. 432-438, http://dx.doi.org/10.1107/S0907444903028956.; McCoy et al., 2005McCoy, A. J.; Grosse-Kunstleve, R. W.; Storoni, L. C. and Read, R. J. (2005). "Likelihood-enhanced fast translation functions". Acta Crystallographica, D61, pp. 458-464, http://dx.doi.org/10.1107/S0907444905001617.) fast and yet able to better model the differences and limitations of the known template has decisively increased its radius of convergence.
Molecular replacement may use any source of coordinates or electron density model. Although traditionally templates derived from other crystallographic studies were most successful, structures determined by NMR in solution or low resolution electron density maps and even low angle scattering (SAXS) envelopes have been successfully exploited in molecular replacement. Recent improvements in the field of cryo-electron microscopy (cryo-EM) have yielded structural information at resolutions around 3.5 Å, approaching low-resolution X-ray crystallography (Amunts et al., 2014Amunts, A.; Brown, A.; Bai, X. C.; Llácer, J. L.; Hussain, T.; Emsley, P.; Long, F.; Murshudov, G.; Scheres, S. H. and Ramakrishnan, V. (2014). "Structure of the yeast mitochondrial large ribosomal subunit". Science, 343, pp. 1485-1489, http://dx.doi.org/10.1126/science.1249410.). As microscopy does provide a direct image at lower spatial resolution, tighter symbiosis between both fields should prove very advantageous. Another extremely interesting development, the use of modeled structures for molecular replacement has been shown to be feasible (Qian et al., 2007Qian, B.; Raman, S.; Das, R.; Bradley, P.; McCoy, A. J.; Read, R. J. and Baker, D. (2007). "High-resolution structure prediction and the crystallographic phase problem". Nature, 450, pp. 259-64, http://dx.doi.org/10.1038/nature06249.). Modeling can be applied to increase the convergence when starting from a poor template from a distant homolog (DiMaio et al., 2011DiMaio, F.; Terwilliger, T. C.; Read, R. J.; Wlodawer, A.; Oberdorfer, G.; Wagner, U.; Valkov, E.; Alon, A.; Fass, D.; Axelrod, H. L.; Das, D.; Vorobiev, S. M.; Iwai, H.; Pokkuluri, P. R. and Baker, D. (2011). "Improved molecular replacement by density- and energy-guided protein structure optimization". Nature, 473, pp. 540-543, http://dx.doi.org/10.1038/nature09964.). Furthermore if de novo structure prediction can reach enough accuracy, this approach provides an ab initio method (Bibby et al., 2012Bibby, J.; Keegan, R. M.; Mayans, O.; Winn, M. D. and Rigden, D. J. (2012). "AMPLE: a cluster-and-truncate approach to solve the crystal structures of small proteins using rapidly computed ab initio models". Acta Crystallographica, D68, pp. 1622-1631, http://dx.doi.org/10.1107/S0907444912039194.).
MACROMOLECULAR AB INITIO PHASING
Top
As previously mentioned, two fundamental barriers hinder direct, ab initio solution (from the native data alone and without previous particular structural knowledge) of the phase problem in the case of macromolecular structures: the larger size of the structure to be determined, leading to a more complex problem where a larger number of parameters need to be determined simultaneously, and the more limited resolution to which macromolecular crystals tend to diffract. In the early 90’s a breakthrough was achieved in the extension of direct methods to small macromolecules diffracting to atomic resolution. Dual-space recycling methods (Miller et al., 1993Miller, R.; DeTitta, G. T.; Jones, R.; Langs, D. A.; Weeks, C. M. and Hauptman, H. A. (1993). "On the application of the minimal principle to solve unknown structures". Science, 259, pp. 1430-1433, http://dx.doi.org/10.1126/science.8451639.; Sheldrick, and Gould, 1995Sheldrick, G. M. and Gould, R. (1995). "Structure solution by iterative peaklist optimization and tangent expansion in space group P1". Acta Crystallographica, B51, pp. 423-431, http://dx.doi.org/10.1107/S0108768195003661.), by iteratively enforcing atomicity in real and reciprocal space, succeeded in constraining a few out of a large pool of trials starting from random atoms into correct, recognizable solution. Atoms are placed randomly but at suitable distances in the unit cell. From them, phases are calculated and subsequently refined according to the direct methods relationships. The new phases are used to calculate an electron density map and its maxima are interpreted as new atom sites. Iterating this procedure may succeed in producing a correct structure, which will be mathematically distinguishable (DeTitta et al., 1994DeTitta, G. T.; Weeks, C. M.; Thuman, P.; Miller, R. and Hauptman, H. A. (1994). "Structure solution by minimal function phase refinement and Fourier filtering: theoretical basis". Acta Crystallographica, A50, pp. 203-210, http://dx.doi.org/10.1107/S0108767393008980.; Fujinaga and Read, 1987Fujinaga, M. and Read, R. J. (1987). "Experiences with a new translation-function program". Journal of Applied Crystallography, 20, pp. 517-521, http://dx.doi.org/10.1107/S0021889887086102.) from the results produced in failed trials. Equal atom structures of up to one thousand independent atoms were first solved by these methods. The size limit could be extended to a few thousand atoms in the favorable cases where heavier atoms, such as inherent iron, would be contained in the structure. Notably, a number of the cases corresponded to particular classes of compounds, such as antibiotics or cyclodextrines, too large to be phased by small molecule methods but for which neither related models for molecular replacement were known, nor derivatizing techniques suitable for proteins would be applicable (Sheldrick et al., 2011Sheldrick, G. M.; Gilmore, C. J.; Hauptman, H. A.; Weeks, C. M.; Miller, R. and Usón, I. (2011). "Ab initio phasing". In Arnold, E.; Himmel, D. M. and Rossmann, M. G. (eds.), International Tables for Crystallography, pp. 413-429. Dordrecht: Kluwer Academic Publishers.). For instance, the glycopeptid antibiotic vancomycin had been crystallized for over 20 years before it could be finally phased by dual-space methods (Schäfer, Schneider and Sheldrick, 1996Schäfer, M.; Schneider, T. R. and Sheldrick, G. M. (1996). "Crystal structure of vancomycin". Structure, 4, pp. 1509-1515, http://dx.doi.org/10.1016/S0969-2126(96)00156-6.; Loll et al., 1997Loll, P. J.; Bevivino, A. E.; Korty, B. D. and Axelsen, P. H. (1997). "Simultaneous Recognition of a Carboxylate-containing Ligand and an Intramolecular Surrogate Ligand in the Crystal Structure of an Asymmetric Vancomycin Dimer". Journal of the American Chemical Society, 119, pp. 1516-1522, http://dx.doi.org/10.1021/ja963566p.).
The exceptionally high quality of the data required for dual-space recycling methods to succeed would have limited their use, as in such cases all alternative phasing approaches are also favored. At extremely high resolution, even sophisticated molecular replacement search using single atoms as models may succeed in solving macromolecular structures (McCoy et al., 2007McCoy, A. J.; Grosse-Kunstleve, R. W.; Adams, P. D.; Winn, M. D.; Storoni, L. C. and Read, R. J. (2007). "Phaser crystallographic software". Journal of Applied Crystallography, 40, pp. 658-674, http://dx.doi.org/10.1107/S0021889807021206.). In practice, dual-space recycling methods became essential for experimental phasing (MIR, MAD, SIR, SAD, SIRAS, RIP) as they succeed in determining large substructures of heavy atoms or anomalous scatterers from noisier data, where conventional direct or Patterson methods fail (Weeks et al., 2003Weeks, C. M.; Adams, P. D.; Berendzen, J.; Brünger, A. T.; Dodson, E. J.; Grosse-Kunstleve, R. W.; Schneider, T. R.; Sheldrick, G. M.; Terwilliger, T. C.; Turkenburg, M. and Usón, I. (2003). "Automatic Solution of Heavy-Atom Substructures". Methods in Enzymology, 374, pp. 37-83, http://dx.doi.org/10.1016/S0076-6879(03)74003-8.). Upcoming, molecular replacement likelihood based methods are also bound to play a central role in the determination of substructures for experimental phasing (Buncóczi et al., 2014Bunkóczi, G.; McCoy, A. J.; Echols, N.; Grosse-Kunstleve, R. W.; Adams, P. D.; Holton, J. M.; Read, R. J. and Terwilliger, T. C. (2014). "Macromolecular X-ray structure determination using weak, single-wavelength anomalous data". Nature Methods, 12, pp. 127-130, http://dx.doi.org/10.1038/nmeth.3212.).
To make up for the lack of atomic resolution data, ab initio phasing required additional constraints beyond atomicity. Sophisticated use of the Patterson function has been exploited (Caliandro et al., 2008Caliandro, R.; Carrozzini, B.; Cascarano, G. L.; De Caro, L.; Giacovazzo, C.; Mazzone, A. and Siliqi, D. (2008). "Crystal structure solution of small-to-medium-sized molecules at non-atomic resolution". Journal of Applied Crystallography, 41, pp. 548-553, http://dx.doi.org/10.1107/S002188980800945X.), as well as extrapolation to include non-measured reflections beyond the diffraction limit was introduced by Giacovazzo (Caliandro et al., 2005aCaliandro, R.; Carrozzini, B.; Cascarano, G. L.; De Caro, L.; Giacovazzo, C. and Siliqi, D. (2005a). "Phasing at resolution higher than the experimental resolution". Acta Crystallographica, D61, pp. 556-565, http://dx.doi.org/10.1107/S090744490500404X.) to improve the experimental data when these conditions were not fulfilled and its use was incorporated to the ab initio phasing case (Caliandro et al., 2005bCaliandro, R.; Carrozzini, B.; Cascarano, G. L.; De Caro, L.; Giacovazzo, C. and Siliqi, D. (2005b). "Ab initio phasing at resolution higher than experimental resolution". Acta Crystallographica, D61, pp. 1080-1087, http://dx.doi.org/10.1107/S0907444905015519.).
Density modification algorithms appropriate for the high, yet not atomic, resolution cases were more efficient than interpreting the map in terms of atoms (Jia-Xing et al., 2005Jia-Xing, Y.; Woolfson, M. M.; Wilson, K. S. and Dodson, E. J. (2005). "A modified ACORN to solve protein structures at resolutions of 1.7 Å or better". Acta Crystallographica, D61, pp. 1465-1475, http://dx.doi.org/10.1107/S090744490502576X.; Sheldrick, 2010Sheldrick, G. M. (2010). "Experimental phasing with SHELXC/D/E: combining chain tracing with density modification". Acta Crystallographica, D66, pp. 479-485, http://dx.doi.org/10.1107/S0907444909038360.; Burla et al., 2012Burla, M. C.; Carrozzini, B.; Cascarano, G. L.; Giacovazzo, C. and Polidori, G. (2012). "VLD algorithm and hybrid Fourier syntheses". Journal of Applied Crystallography, 45, pp. 1287-1294, http://dx.doi.org/10.1107/S0021889812041155.).
Tighter stereochemical constraints than atomicity were introduced to extend ab initio phasing to non-atomic resolution. The search of small secondary structure predicted helical fragments (Glykos and Kokkinidis, 2003Glykos, N. M. and Kokkinidis, M. (2003). "Structure determination of a small protein through a 23-dimensional molecular-replacement search". Acta Crystallographica, D59, pp. 709-718, http://dx.doi.org/10.1107/S0907444903002889.) or nucleic acid bases (Robertson and Scott, 2008Robertson, M. P. and Scott, W. G. (2008). "A general method for phasing novel complex RNA crystal structures without heavy-atom derivatives". Acta Crystallographica, D64, pp. 738-744, http://dx.doi.org/10.1107/S0907444908011578.) led to solve some structures. The approach implemented in ARCIMBOLDO relies on the combination of locating model fragments such as polyalanine alpha-helices through maximum-likelihood molecular replacement and density modification. Given the difficulties to discriminate correct small substructures, many possible fragment substructures are tested in parallel, in a grid or supercomputer (Rodríguez, et al., 2009Rodríguez, D.; Grosse, C.; Himmel, S.; González, C.; Martínez de Ilarduya, I.; Becker, S.; Sheldrick, G. M. and Usón, I. (2009). "Crystallographic ab initio protein solution below atomic resolution". Nature Methods, 6, pp. 651-653., Proepper, et al., 2014Proepper, K.; Meindl, K.; Sammito, M.; Dittrich, B.; Sheldrick, G. M.; Pohl, E. and Uson, I. (2014). "Structure solution of DNA-binding proteins and complexes with ARCIMBOLDO libraries". Acta Crystallographica, D70, pp. 1743-1757, http://dx.doi.org/10.1107/S1399004714007603.). The method has been named after the Italian painter Arcimboldo, who used to compose portraits out of fruits and vegetables. In the case of the program, most collections of fragments remain a “still-life”, but some are correct enough for density modification and main chain tracing to reveal the protein’s true portrait. Beyond alpha-helices, other fragments can be exploited in analogous way: libraries of helices with modeled side-chains, beta strands, predictable fragments such as DNA-binding folds or fragments selected from distant homologs up to libraries of small local folds that are used to enforce non-specific tertiary structure, thus restoring the ab initio nature of the method (Sammito et al., 2013Sammito, M. D.; Millán, C.; Rodríguez, D.; M. de Ilarduya, I.; Meindl, K.; De Marino, I.; Petrillo, G.; Buey, R. M.; de Pereda, J. M.; Zeth, K.; Sheldrick, G. M. and Usón, I. (2013). "Exploiting tertiary structure through local folds for crystallographic phasing". Nature Methods, 10, pp. 1099-1101, http://dx.doi.org/10.1038/nmeth.2644.). Using these methods, a number of unknown macromolecules with a few thousand atoms and resolutions around 2 Å have been solved.
ACKOWLEDGEMENTSTop
This work was supported by grants BFU2012-35367 and BIO2013-49604- EXP (the Spanish MINECO) and Generalitat de Catalunya (2014SGR-997). We thank Ehmke Pohl for helpful discussion and corrections.
REFERENCESTop
|
| ○ | Abad-Zapatero, C.; Abdel-Meguid, S. S.; Johnson, J. E.; Leslie, A. G.; Rayment, I.; Rossmann, M. G.; Suck, D. and Tsukihara, T. (1980). "Structure of southern bean mosaic virus at 2.8Å resolution". Nature, 286, pp. 33-39. http://dx.doi.org/10.1038/286033a0 |
| ○ | Adams, M. J.; Blundell, T. L.; Dodson, E. J.; Dodson, G. G.; Vijayan, M.; Baker, E. N.; Harding, M. M.; Hodgkin, D. C.; Rimmer, B. and Sheat, S. (1969). "Structure of Rhombohedral 2 Zinc Insulin Crystals". Nature, 224, pp. 491. http://dx.doi.org/10.1038/224491a0 |
| ○ | Allen, F. H. (2002). "The Cambridge Structural Database: a quarter of a million crystal structures and rising". Acta Crystallographica, B58, pp. 380-388. http://dx.doi.org/10.1107/S0108768102003890 |
| ○ | Amunts, A.; Brown, A.; Bai, X. C.; Llácer, J. L.; Hussain, T.; Emsley, P.; Long, F.; Murshudov, G.; Scheres, S. H. and Ramakrishnan, V. (2014). "Structure of the yeast mitochondrial large ribosomal subunit". Science, 343, pp. 1485-1489. http://dx.doi.org/10.1126/science.1249410 |
| ○ | Armstrong, H. E. (1927). "Poor Common Salt!". Nature, 120, pp. 478. http://dx.doi.org/10.1038/120478b0 |
| ○ | Astbury, W. T. and Street, A. (1932). "A. X-Ray studies of the structure of hair, wool, and related fibers. I. General". Philosophical Transactions of the Royal Society A, 230, pp. 75-101. http://dx.doi.org/10.1098/rsta.1932.0003 |
| ○ | Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N. and Bourne, P. E. (2000). "The Protein Data Bank". Nucleic Acids Research, 28, pp. 235-242. http://dx.doi.org/10.1093/nar/28.1.235 |
| ○ | Bernal, J. D. and Crowfoot, D. (1934). "X-Ray Photographs of Crystalline Pepsin". Nature, 133, pp. 794-795. http://dx.doi.org/10.1038/133794b0 |
| ○ | Bernstein, F. C.; Koetzle, T. F.; Williams, G. J. B.; Meyer, E. F.; Brice, M. D.; Rodgers, J. R.; Kennard, O.; Shimanouchi, T. and Tasumi, M. (1977). "The Protein Data Bank: a computer-based archival file for macromolecular structures". Journal of Molecular Biology, 112, pp. 535-542. http://dx.doi.org/10.1016/S0022-2836(77)80200-3 |
| ○ | Bibby, J.; Keegan, R. M.; Mayans, O.; Winn, M. D. and Rigden, D. J. (2012). "AMPLE: a cluster-and-truncate approach to solve the crystal structures of small proteins using rapidly computed ab initio models". Acta Crystallographica, D68, pp. 1622-1631. http://dx.doi.org/10.1107/S0907444912039194 |
| ○ | Bragg, W. L. (1913a). "The Diffraction of Short Electromagnetic Waves by a Crystal". Mathematical Proceedings of the Cambridge Philosophical Society, 17, pp. 43-57. |
| ○ | Bragg, W. L. (1913b). "The Structure of Some Crystals as indicated by their Diffraction of X-rays". Proceedings of the Royal Society of London, 89, pp. 248-277. http://dx.doi.org/10.1098/rspa.1913.0083 |
| ○ | Bragg, W. L. (1925). "The Crystalline Structure of Inorganic Salts". Nature, 116, pp. 557. http://dx.doi.org/10.1038/116249a0 |
| ○ | Bragg, W. H. and Bragg, W. L. (1913a). "The Reflexion of X-rays by Crystals". Proceedings of the Royal Society of London A, 88 (605), pp. 428-438. http://dx.doi.org/10.1098/rspa.1913.0040 |
| ○ | Bragg, W. H. and Bragg, W. L. (1913b). "The structure of the diamond". Nature, 91, pp. 557. http://dx.doi.org/10.1038/091557a0 |
| ○ | Broenniman, C.; Eikenberry, E. F.; Henrich, B.; Horisberger, G.; Huelsen, G.; Pohl, E.; Schmitt, B.; Schulze-Briese, C.; Suzuki, M.; Tomizaki, T.; Toyokawa, A. and Wagner, A. (2006). "The Pilatus 1M detector". Journal of Synchrotron Radiation, 13, pp. 120-133. http://dx.doi.org/10.1107/S0909049505038665 |
| ○ | Bunkóczi, G.; McCoy, A. J.; Echols, N.; Grosse-Kunstleve, R. W.; Adams, P. D.; Holton, J. M.; Read, R. J. and Terwilliger, T. C. (2014). "Macromolecular X-ray structure determination using weak, single-wavelength anomalous data". Nature Methods, 12, pp. 127-130. http://dx.doi.org/10.1038/nmeth.3212 |
| ○ | Burla, M. C.; Carrozzini, B.; Cascarano, G. L.; Giacovazzo, C. and Polidori, G. (2012). "VLD algorithm and hybrid Fourier syntheses". Journal of Applied Crystallography, 45, pp. 1287-1294. http://dx.doi.org/10.1107/S0021889812041155 |
| ○ | Caliandro, R.; Carrozzini, B.; Cascarano, G. L.; De Caro, L.; Giacovazzo, C. and Siliqi, D. (2005a). "Phasing at resolution higher than the experimental resolution". Acta Crystallographica, D61, pp. 556-565. http://dx.doi.org/10.1107/S090744490500404X |
| ○ | Caliandro, R.; Carrozzini, B.; Cascarano, G. L.; De Caro, L.; Giacovazzo, C. and Siliqi, D. (2005b). "Ab initio phasing at resolution higher than experimental resolution". Acta Crystallographica, D61, pp. 1080-1087. http://dx.doi.org/10.1107/S0907444905015519 |
| ○ | Caliandro, R.; Carrozzini, B.; Cascarano, G. L.; De Caro, L.; Giacovazzo, C.; Mazzone, A. and Siliqi, D. (2008). "Crystal structure solution of small-to-medium-sized molecules at non-atomic resolution". Journal of Applied Crystallography, 41, pp. 548-553. http://dx.doi.org/10.1107/S002188980800945X |
| ○ | Champness, J. N.; Bloomer, A. C.; Bricogne, G.; Butler, P. G. and Klug, A. (1976). "The structure of the protein disk of tobacco mosaic virus to 5A resolution". Nature, 259, pp. 20-24. http://dx.doi.org/10.1038/259020a0 |
| ○ | Cowtan, K. D. and Main, P. (1993). "Improvement of macromolecular electron-density maps by the simultaneous application of real and reciprocal space constraints". Acta Crystallographica, D49, pp. 148-157. http://dx.doi.org/10.1107/S0907444992007698 |
| ○ | Crowfoot, D.; Bunn, C. W.; Rogers-Low, B. W. and Turner-Jones, A. (1949). "X-ray crystallographic investigation of the structure of penicillin". In Clarke, H. T.; Johnson, J. R. and Robinson, R. (eds.), Chemistry of Penicillin, pp. 310-367. Princeton University Press. |
| ○ | Crowther, R. A. and Blow, D. W. (1967). "A Method of Positioning a Known Molecule in an Unknown Crystal Structure". Acta Crystallographica, 23, pp. 544-548. http://dx.doi.org/10.1107/S0365110X67003172 |
| ○ | Dauter, Z. and Adamiak, D. A. (2001). "Anomalous signal of phosphorus used for phasing DNA oligomer: importance of data redundancy". Acta Crystallographica, D57, pp. 990-995. http://dx.doi.org/10.1107/S0907444901006382 |
| ○ | Dauter, Z.; Dauter, M.; de La Fortelle, E.; Bricogne, G. and Sheldrick, G. M. (1999). "Can anomalous signal of sulfur become a tool for solving protein crystal structures?". Journal of Molecular Biology, 289, pp. 83-92. http://dx.doi.org/10.1006/jmbi.1999.2743 |
| ○ | DeTitta, G. T.; Weeks, C. M.; Thuman, P.; Miller, R. and Hauptman, H. A. (1994). "Structure solution by minimal function phase refinement and Fourier filtering: theoretical basis". Acta Crystallographica, A50, pp. 203-210. http://dx.doi.org/10.1107/S0108767393008980 |
| ○ | DiMaio, F.; Terwilliger, T. C.; Read, R. J.; Wlodawer, A.; Oberdorfer, G.; Wagner, U.; Valkov, E.; Alon, A.; Fass, D.; Axelrod, H. L.; Das, D.; Vorobiev, S. M.; Iwai, H.; Pokkuluri, P. R. and Baker, D. (2011). "Improved molecular replacement by density- and energy-guided protein structure optimization". Nature, 473, pp. 540-543. http://dx.doi.org/10.1038/nature09964 |
| ○ | Ewald, P. P. (1913). "About the theory of the interference of X-rays in crystals (Zur Theorie der interferenzen der Röntgen-strahlen in kristallen)". Physikalische Zeitschrift, 14, pp. 465-472. |
| ○ | Franklin, R. and Gosling, R. G. (1953). "Molecular Configuration in Sodium Thymonucleate". Nature, 171, pp. 740-741. http://dx.doi.org/10.1038/171740a0 |
| ○ | Friedrich, W.; Knipping, P. and Laue, M. (1912). "Interferenz-Erscheinungen bei Röntgenstrahlen". Sitzungsberichte der Königlich Bayerische Akademie der Wissenschaften, pp. 303-322. |
| ○ | Fujinaga, M. and Read, R. J. (1987). "Experiences with a new translation-function program". Journal of Applied Crystallography, 20, pp. 517-521. http://dx.doi.org/10.1107/S0021889887086102 |
| ○ | Garman, E. F. and Schneider, T. R. (1997). "Macromolecular Cryocrystallography". Journal of Applied Crystallography, 30, pp. 211-237. http://dx.doi.org/10.1107/S0021889897002677 |
| ○ | Giacovazzo, C. (2011). Fundamentals of crystallography (3rd ed). Oxford, New York: Oxford University Press - International Union of Crystallography texts on crystallography. http://dx.doi.org/10.1093/acprof:oso/9780199573653.001.0001 |
| ○ | Glykos, N. M. and Kokkinidis, M. (2003). "Structure determination of a small protein through a 23-dimensional molecular-replacement search". Acta Crystallographica, D59, pp. 709-718. http://dx.doi.org/10.1107/S0907444903002889 |
| ○ | Green, D. W.; Ingram, V. M. and Perutz, M. F. (1954). "Structure of haemoglobin: IV. Sign determination by the isomorphous replacement method". Proceedings of the Royal Society of London. Series A, 225, pp. 287-307. http://dx.doi.org/10.1098/rspa.1954.0203 |
| ○ | Hauptman, H. and Karle, J. (1953). Solution of the phase problem I. The centrosymmetric crystal. Dayton, Ohio: American Crystallographic Association. |
| ○ | Harker, D. (1936). "The application of the three-dimensional Patterson method and the crystal structures of proustite, Ag3AsS3, and pyrargyrite, Ag3SbS3". The Journal of Chemical Physics, 4, pp. 381-390. http://dx.doi.org/10.1098/rspa.1954.0203 |
| ○ | Harker, D. (1956). "The determination of the phases of the structure factors of non-centrosymmetric crystals by the method of double isomorphous replacement". Acta Crystallographica, 9, pp. 1-9. http://dx.doi.org/10.1107/S0365110X56000012 |
| ○ | Harker, D. and Kasper, J. S. (1948). "Phases of Fourier coefficients directly from crystal diffraction data". Acta Crystallographica, 1, pp. 70-75. http://dx.doi.org/10.1107/S0365110X4800020X |
| ○ | Harrison, S. C.; Olson, A. J.; Schutt, C. E.; Winkler, F. K. and Bricogne, G. (1978). "Tomato bushy stunt virus at 2.9 Å resolution". Nature, 276, pp. 368-373. http://dx.doi.org/10.1038/276368a0 |
| ○ | Hendrickson, W. A. (1991). "Determination of Macromolecular Structures from Anomalous Diffraction of Synchrotron Radiation". Science, 254, pp. 51-58. http://dx.doi.org/10.1126/science.1925561 |
| ○ | Hendrickson, W. A. and Teeter, M. M. (1981). "Structure of the hydrophobic protein crambin determined directly from the anomalous scattering of sulphur". Nature, 290, pp. 107-113. http://dx.doi.org/10.1038/290107a0 |
| ○ | Hodgkin, D. C.; Kamper, J.; Mackay, M.; Pickworth, J.; Trueblood, K. N. and White, J. G. (1956). "Structure of vitamin B-12". Nature, 178, pp. 64-66. http://dx.doi.org/10.1038/178064a0 |
| ○ | Hope, H. (1990). "Crystallography of biological macromolecules at ultra-low temperature". Annual Review of Biophysics and Biophysical Chemistry, 19, pp. 107-126. http://dx.doi.org/10.1146/annurev.bb.19.060190.000543 |
| ○ | Huber, R. (1965). "Die automatisierte Faltmolekülmethode". Acta Crystallographica, 19, pp. 353-356. http://dx.doi.org/10.1107/S0365110X65003444 |
| ○ | Jia-Xing, Y.; Woolfson, M. M.; Wilson, K. S. and Dodson, E. J. (2005). "A modified ACORN to solve protein structures at resolutions of 1.7 Å or better". Acta Crystallographica, D61, pp. 1465-1475. http://dx.doi.org/10.1107/S090744490502576X |
| ○ | Karle, J. and Hauptman, H. (1950). "The phases and magnitudes of the structure factors". Acta Crystallographica, 3, pp. 181-187. http://dx.doi.org/10.1107/S0365110X50000446 |
| ○ | Karle, J. and Hauptman, H. (1956). "A theory of phase determination for the four types of non-centrosymmetric space groups 1P222, 2P22, 3P12, 3P22". Acta Crystallographica, 9, pp. 635-651. http://dx.doi.org/10.1107/S0365110X56001741 |
| ○ | Kendrew, J. C.; Bodo, G.; Dintzis, H. M.; Parrish, R. G.; Wyckoff, H. and Phillips, D. C. (1958). "A three dimensional model of the myoglobin molecule obtained by X-ray analysis". Nature, 181, pp. 662-666. http://dx.doi.org/10.1038/181662a0 |
| ○ | Kissinger, C. L.; Gehlhaar, D. K. and Fogel, D. B. (1999). "Rapid automated molecular replacement by evolutionary search". Acta Crystallographica, D55, pp. 484-491. http://dx.doi.org/10.1107/S0907444998012517 |
| ○ | von Laue, M. (1912). "Eine quantitative prüfung der theorie für die interferenz-erscheinungen bei Röntgenstrahlen". Sitzungsberichte der Königlich Bayerische Akademie der Wissenschaften, pp. 363-373. |
| ○ | Liu, Q.; Dahmane, T.; Zhang, Z.; Assur, Z.; Brasch, J.; Shapiro, L.; Mancia, F. and Hendrickson, W. A. (2012). "Structures from Anomalous Diffraction of Native Biological Macromolecules". Science, 336, pp. 1033-1037. http://dx.doi.org/10.1126/science.1218753 |
| ○ | Loll, P. J.; Bevivino, A. E.; Korty, B. D. and Axelsen, P. H. (1997). "Simultaneous Recognition of a Carboxylate-containing Ligand and an Intramolecular Surrogate Ligand in the Crystal Structure of an Asymmetric Vancomycin Dimer". Journal of the American Chemical Society, 119, pp. 1516-1522. http://dx.doi.org/10.1021/ja963566p |
| ○ | McCoy, A. J.; Grosse-Kunstleve, R. W.; Storoni, L. C. and Read, R. J. (2005). "Likelihood-enhanced fast translation functions". Acta Crystallographica, D61, pp. 458-464. http://dx.doi.org/10.1107/S0907444905001617 |
| ○ | McCoy, A. J.; Grosse-Kunstleve, R. W.; Adams, P. D.; Winn, M. D.; Storoni, L. C. and Read, R. J. (2007). "Phaser crystallographic software". Journal of Applied Crystallography, 40, pp. 658-674. http://dx.doi.org/10.1107/S0021889807021206 |
| ○ | Miller, R.; DeTitta, G. T.; Jones, R.; Langs, D. A.; Weeks, C. M. and Hauptman, H. A. (1993). "On the application of the minimal principle to solve unknown structures". Science, 259, pp. 1430-1433. http://dx.doi.org/10.1126/science.8451639 |
| ○ | Mueller, M.; Wang, M. and Schulze-Briese, C. (2012). "Optimal fine ϕ-slicing for single-photon-counting pixel detectors". Acta Crystallographica, D68, pp. 42-56. http://dx.doi.org/10.1107/S0907444911049833 |
| ○ | Oszlanyi, G. and Süto, A. (2004). "Ab initio structure solution by charge flipping". Acta Crystallographica, A60, pp. 134-141. http://dx.doi.org/10.1107/S0108767303027569 |
| ○ | Patterson, A. L. (1935). "A direct method for the determination of the components of interatomic distances in crystals". Zeitschrift für Kristallographie, 90, pp. 517-542. http://dx.doi.org/10.1524/zkri.1935.90.1.517 |
| ○ | Pauling, L. and Corey, R. B. (1951). "The pleated sheet, a new layer configuration of polypeptide chains". Proceedings of the National Academy of Sciences of the USA, 37, pp. 251-256. http://dx.doi.org/10.1073/pnas.37.5.251 |
| ○ | Pauling, L.; Corey, R. B. and Branson, H. R. (1951). "The structure of proteins, two hydrogen-bonded helical configurations of the polypeptide chain". Proceedings of the National Academy of Sciences of the USA, 37, pp. 205-211. http://dx.doi.org/10.1073/pnas.37.4.205 |
| ○ | Proepper, K.; Meindl, K.; Sammito, M.; Dittrich, B.; Sheldrick, G. M.; Pohl, E. and Uson, I. (2014). "Structure solution of DNA-binding proteins and complexes with ARCIMBOLDO libraries". Acta Crystallographica, D70, pp. 1743-1757. http://dx.doi.org/10.1107/S1399004714007603 |
| ○ | Qian, B.; Raman, S.; Das, R.; Bradley, P.; McCoy, A. J.; Read, R. J. and Baker, D. (2007). "High-resolution structure prediction and the crystallographic phase problem". Nature, 450, pp. 259-64. http://dx.doi.org/10.1038/nature06249 |
| ○ | Robertson, M. P. and Scott, W. G. (2008). "A general method for phasing novel complex RNA crystal structures without heavy-atom derivatives". Acta Crystallographica, D64, pp. 738-744. http://dx.doi.org/10.1107/S0907444908011578 |
| ○ | Rodríguez, D.; Grosse, C.; Himmel, S.; González, C.; Martínez de Ilarduya, I.; Becker, S.; Sheldrick, G. M. and Usón, I. (2009). "Crystallographic ab initio protein solution below atomic resolution". Nature Methods, 6, pp. 651-653. |
| ○ | Rossmann, M. G. (1972). The Molecular Replacement Method. New York: Gordon & Breach. |
| ○ | Rossmann, M. G. and Blow, D. M. (1962). "The Detection of Subunits within the Crystallographic Asymmetric Unit". Acta Crystallographica, 15, pp. 24-31. http://dx.doi.org/10.1107/S0365110X62000067 |
| ○ | Sammito, M. D.; Millán, C.; Rodríguez, D.; M. de Ilarduya, I.; Meindl, K.; De Marino, I.; Petrillo, G.; Buey, R. M.; de Pereda, J. M.; Zeth, K.; Sheldrick, G. M. and Usón, I. (2013). "Exploiting tertiary structure through local folds for crystallographic phasing". Nature Methods, 10, pp. 1099-1101. http://dx.doi.org/10.1038/nmeth.2644 |
| ○ | Sayre, D. (1952). "The squaring method: a new method for phase determination". Acta Crystallographica, 5, pp. 60-65. http://dx.doi.org/10.1107/S0365110X52000137 |
| ○ | Schäfer, M.; Schneider, T. R. and Sheldrick, G. M. (1996). "Crystal structure of vancomycin". Structure, 4, pp. 1509-1515. http://dx.doi.org/10.1016/S0969-2126(96)00156-6 |
| ○ | Sheldrick, G. M. and Gould, R. (1995). "Structure solution by iterative peaklist optimization and tangent expansion in space group P1". Acta Crystallographica, B51, pp. 423-431. http://dx.doi.org/10.1107/S0108768195003661 |
| ○ | Sheldrick, G. M. (2008). "A short history of SHEL". Acta Crystallographica, A64, pp. 112-122. http://dx.doi.org/10.1107/S0108767307043930 |
| ○ | Sheldrick, G. M. (2010). "Experimental phasing with SHELXC/D/E: combining chain tracing with density modification". Acta Crystallographica, D66, pp. 479-485. http://dx.doi.org/10.1107/S0907444909038360 |
| ○ | Sheldrick, G. M.; Gilmore, C. J.; Hauptman, H. A.; Weeks, C. M.; Miller, R. and Usón, I. (2011). "Ab initio phasing". In Arnold, E.; Himmel, D. M. and Rossmann, M. G. (eds.), International Tables for Crystallography, pp. 413-429. Dordrecht: Kluwer Academic Publishers. |
| ○ | Storoni, L. C.; McCoy, A. J. and Read, R. J. (2004). "Likelihood-enhanced fast rotation functions". Acta Crystallographica, D60, pp. 432-438. http://dx.doi.org/10.1107/S0907444903028956 |
| ○ | Wang, B.-C. (1985). "Resolution of phase ambiguity in macromolecular crystallography". Methods in Enzymology, 115, pp. 90-112. http://dx.doi.org/10.1016/0076-6879(85)15009-3 |
| ○ | Wang, A. H.-J.; Quigley, G. J.; Kolpak, F. J.; Crawford, J. L.; van Boom, J. H.; van der Marel, G. and Rich, A. (1979). "Molecular structure of a left-handed double helical DNA fragment at atomic resolution". Nature, 282, pp. 680-686. http://dx.doi.org/10.1038/282680a0 |
| ○ | Watson, J. D. and Crick, F. H. C. (1953). "Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid". Nature, 171, pp. 737-738. http://dx.doi.org/10.1038/171737a0 |
| ○ | Weeks, C. M.; Adams, P. D.; Berendzen, J.; Brünger, A. T.; Dodson, E. J.; Grosse-Kunstleve, R. W.; Schneider, T. R.; Sheldrick, G. M.; Terwilliger, T. C.; Turkenburg, M. and Usón, I. (2003). "Automatic Solution of Heavy-Atom Substructures". Methods in Enzymology, 374, pp. 37-83. http://dx.doi.org/10.1016/S0076-6879(03)74003-8 |
| ○ | Weinert, T.; Olieric, V.; Waltersperger, S.; Panepucci, E.; Chen, L.; Zhang, H.; Zhou, D.; Rose, J.; Ebihara, A.; Kuramitsu, S.; Li, D.; Howe, N.; Schnapp, G.; Pautsch, A.; Bargsten, K.; Prota, A. E.; Surana, P.; Kottur, J.; Nair, D. T.; Basilico, F.; Cecatiello, V.; Pasqualato, S.; Boland, A.; Weichenrieder, O.; Wang, B.-C.; Steinmetz, M. O.; Caffrey, M. and Wang, M. (2015). "Fast native-SAD phasing for routine macromolecular structure determination". Nature Methods, 12, pp. 131-133. http://dx.doi.org/10.1038/nmeth.3211 |
| ○ | Yang, C.; Pflugrath, J. W.; Courville, D. A.; Stence, C. N. and Ferrara, J. D. (2003). "Away from the edge: SAD phasing from the sulfur anomalous signal measured in-house with chromium radiation". Acta Crystallographica, D59, pp. 1943-1957. http://dx.doi.org/10.1107/S0907444903018547 |
|