MEDICINA, CIENCIA Y REALIDAD DE LAS ENFERMEDADES RARAS / MEDICINE, SCIENCE AND REALITY OF RARE DISEASES
LAS ENFERMEDADES RARAS EN LAS PATOLOGÍAS NEUROMETABÓLICAS
Julio Montoya
Universidad de Zaragoza / Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) / Instituto de Investigación Sanitaria de Aragón
ORCID iD: http://orcid.org/0000-0003-1770-6299
jmontoya@unizar.es
Joaquín Arenas
Hospital 12 de Octubre (i+12) / Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER)
ORCID iD: http://orcid.org/0000-0002-2877-5049
Joaquin.arenas@salud.madrid.org
Eduardo Ruiz-Pesini
Fundación ARAID / Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) / Instituto de Investigación Sanitaria de Aragón
ORCID iD: http://orcid.org/0000-0002-0269-7337
eduruiz@unizar.es
Miguel A. Martín-Casanueva
Hospital 12 de Octubre (i+12) / Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER)
ORCID iD: http://orcid.org/0000-0003-4741-772X
mamcasanueva.imas12@h12o.es
RESUMEN
Las miopatías metabólicas son un grupo de trastornos genéticos que disminuyen la capacidad del músculo esquelético para utilizar sustratos energéticos y sintetizar ATP. Estas alteraciones pueden clasificarse en tres tipos fundamentalmente: i) trastornos del metabolismo de los carbohidratos (del glucógeno y de la glucosa), ii) defectos del metabolismo lipídico, y iii) alteraciones de la fosforilación oxidativa –OXPHOS-. Las dos primeras se deben a deficiencias enzimáticas de las rutas metabólicas de degradación y síntesis de glúcidos y lípidos y muestran diversas manifestaciones clínicas, pero una buena parte de ellas cursan con intolerancia al ejercicio. Aunque un buen número de pacientes con estos trastornos musculares presentan síntomas en la infancia, el diagnóstico normalmente se retrasa hasta la segunda y tercera década de la vida. Por tanto, reconocer las características clínicas de estas deficiencias conduce a un diagnóstico precoz y a un mejor tratamiento. Las enfermedades mitocondriales son un grupo de trastornos originados por una deficiencia en la síntesis de ATP a través del sistema de fosforilación oxidativa. Este sistema está formado por proteínas codificadas en los dos genomas de la célula (nuclear y mitocondrial) y, por tanto, pueden presentar un modelo de herencia mendeliano o materno. En esta revisión se describirán las características especiales del sistema genético mitocondrial y las principales mutaciones que causan enfermedades en humanos.
RARE DISEASES IN NEUROMETABOLIC PATHOLOGIES
ABSTRACT
Metabolic myopathies are genetic disorders that decrease the capacity of skeletal muscle to use energy substrates and ATP. These disturbances can be classified into three categories: i) disorders of carbohydrate metabolism (glycogen and glucose), ii) defects in lipid metabolism, and iii) dysfunctions of oxidative phosphorylation -OXPHOS-. The first two are caused by enzyme deficiencies involved in the metabolic pathways of degradation and synthesis of carbohydrates and lipids, and although they show distinct clinical manifestations, exercise intolerance is a predominant symptom present in most of them. Although a good number of patients with these muscle disorders display symptoms in childhood, the diagnosis is often delayed until the second and third decades of life. Therefore, recognizing the clinical features of these deficiencies can lead to an earlier diagnosis and better treatment. Mitochondrial diseases are a group of disorders caused by an alteration of the oxidative phosphorylation system, that leads to a deficient synthesis of ATP. This system is composed of proteins codified in the two genetic systems of the cell, the nuclear and mitochondrial genomes, and, therefore, the mode of inheritance could be either Mendelian or maternal. This review will describe the special characteristics of the mitochondrial genetic system and the main mutations in mtDNA that cause human diseases.
Recibido: 30-04-2015; Aceptado: 13-05-2016.
Cómo citar este artículo/Citation: Montoya, J.; Arenas, J.; Ruiz-Pesini, E. y Martín-Casanueva, M.A. (2018). Las enfermedades raras en las patologías neurometabólicas. Arbor, 194 (789): a461. https://doi.org/10.3989/arbor.2018.789n3003
PALABRAS CLAVE: Glucogenosis musculares; miopatías lipídicas; intolerancia al ejercicio; DNA mitocondrial; DNA nuclear; enfermedades mitocondriales; mitocondria; sistema de fosforilación oxidativa.
KEYWORDS: Muscle glycogenosis; lipid storage myopathies; exercise intolerance; muscle mitochondria; mitochondrial diseases; mitochondrial DNA; nuclear DNA; OXPHOS.
Copyright: © 2018 CSIC. Este es un artículo de acceso abierto distribuido bajo los términos de la licencia de uso y distribución Creative Commons Reconocimiento 4.0 Internacional (CC BY 4.0).
CONTENIDOS
1. MIOPATÍAS METABÓLICAS Top
Las miopatías metabólicas son un grupo de trastornos genéticos que disminuyen la capacidad del músculo esquelético para utilizar sustratos energéticos y sintetizar ATP. Estas alteraciones pueden clasificarse en tres tipos fundamentalmente: i) trastornos del metabolismo de los carbohidratos (del glucógeno y de la glucosa), ii) defectos del metabolismo lipídico, y iii) lesiones mitocondriales, aquellas debidas a alteraciones de la fosforilación oxidativa –OXPHOS- (estas últimas serán tratadas en el siguiente apartado específico de este artículo). Estas enfermedades muestran diversas manifestaciones clínicas, pero suelen cursar frecuentemente con intolerancia al ejercicio (IE). Aunque gran parte de los pacientes con miopatía metabólica presentan síntomas de IE en la infancia, el diagnóstico normalmente se retrasa hasta la segunda y tercera década de la vida. Por tanto, reconocer las características clínicas de estas deficiencias conduce a un diagnóstico precoz y un mejor tratamiento de los pacientes.
1.1. Alteraciones de los carbohidratos
1.1.1. Enfermedad de Pompe, déficit de alfa 1,4 glucosidasa ácida (GAA)
Esta proteína es una enzima lisosomal codificada por el gen GAA, cuya deficiencia conlleva una acumulación de glucógeno en el lisosoma, principalmente, del tejido muscular. La incidencia es <1:40.000 nacimientos. Existen presentaciones infantiles, juveniles y del adulto, con diferentes grados de gravedad determinados por el nivel de actividad residual de la enzima, desde el 1% en la forma infantil hasta 10-20% en el adulto. Los niños presentan miocardiopatía hipertrófica, cianosis, macroglosia, hepatomegalia, hipotonía severa y dificultad respiratoria, que suele ser letal en ausencia de tratamiento enzimático sustitutivo (TSE) –myozyme®-, por lo que un diagnóstico precoz es muy importante (Lim, Li y Raben, 2014Lim, J.-A., Li, L. y Raben, N. (2014). Pompe disease: from pathophysiology to therapy and back again. Frontiers in Aging Neuroscience, 6, 177. https://doi.org/10.3389/fnagi.2014.00177.). En el Pompe del adulto se da la afectación muscular sin lesión cardíaca. Actualmente el diagnóstico de confirmación se puede realizar mediante análisis enzimático en linfocitos (más rápido aunque menos específico) y cultivo de fibroblastos, o mediante análisis genético (incluso iniciada la TSE), con el fin de confirmar la enfermedad en niños o en las variantes más tardías (existen más de 450 mutaciones) para su caracterización genética, pues se están estudiando otras terapias como el uso de chaperonas moleculares que funcionan con efectividad según el tipo de mutación presente.
1.1.2. Deficiencia de fosforilasa muscular, miofosforilasa, enfermedad de McArdle, enfermedad de almacenamiento del glucógeno tipo V (GSDV)
Mutaciones en el gen PYGM (>140) provocan la perdida de la actividad de la enzima glucógeno fosforilasa muscular, y por lo tanto un bloqueo completo en la degradación del glucógeno (Figura 1). Cuando se realizan contracciones musculares intensas, se estimula la glucogenolisis anaeróbica y la afectación muscular en estos enfermos. El glucógeno también es necesario para el metabolismo oxidativo, por lo cual la disponibilidad de sustratos para OXPHOS disminuye, disminuyendo también la capacidad para realizar ejercicio de intensidad moderada. En los pacientes aparece el fenómeno second-wind, mediante el cual cuando notan las primeras contracturas y paran el ejercicio, hay una readaptación metabólica consistente en un aumento de captación de glucosa y del catabolismo de ácidos grasos (Haller y Vissing, 2002Haller, R. G. y Vissing, J. (2002). Spontaneous second wind and glucose-induced second second wind in McArdle disease: oxidative mechanisms. Archives of Neurology, 59 (9), pp. 1395-1402. https://doi.org/10.1001/archneur.59.9.1395.). Este hecho ocurre tras 6 a 8 minutos de ejercicio y se asocia con una disminución en la frecuencia cardíaca que es proporcional a un aumento en la capacidad oxidativa del músculo. El esfuerzo máximo, como levantar o empujar objetos pesados, causa rápidamente fatiga y contracturas musculares, mialgia y rabdomiolisis, que puede dar lugar a mioglobinuria (orinas oscuras) en > 50% de los casos; y alrededor del 10% de los pacientes pueden sufrir fracaso renal (Lucía et al. 2012Lucía, A., Ruiz, J. R., Santalla, A., Nogales-Gadea, G., Rubio, J. C., García-Consuegra, I. y Navarro, C. et al. (2012). Genotypic and phenotypic features of McArdle disease: insights from the Spanish national registry. Journal of Neurology, Neurosurgery & Psychiatry, 83 (3), pp. 322-328. https://doi.org/10.1136/jnnp-2011-301593.). Los pacientes experimentan fatiga, taquicardia, y en ocasiones dificultad respiratoria con el ejercicio de más larga duración de intensidad sostenida como caminar cuesta arriba (Sharp y Haller, 2014Sharp, L. J. y Haller, R. G. (2014). Metabolic and mitochondrial myopathies. Neurologic Clinics, 32 (3), pp. 777-799. https://doi.org/10.1016/j.ncl.2014.05.001.). Se produce debilidad muscular fija en un 25% de los pacientes, generalmente en mayores de 40 años. Es la miopatía pura metabólica más frecuente, estimándose una prevalencia en España de 1:170.000 (Lucía et al. 2012Lucía, A., Ruiz, J. R., Santalla, A., Nogales-Gadea, G., Rubio, J. C., García-Consuegra, I. y Navarro, C. et al. (2012). Genotypic and phenotypic features of McArdle disease: insights from the Spanish national registry. Journal of Neurology, Neurosurgery & Psychiatry, 83 (3), pp. 322-328. https://doi.org/10.1136/jnnp-2011-301593.; Quinlivan et al. 2010Quinlivan, R., Buckley, J., James, M., Twist, A., Ball, S., Duno, M. y Winer, J. et al. (2010). McArdle disease: a clinical review. Journal of Neurology, Neurosurgery & Psychiatry, 81 (11), pp. 1182-1188. https://doi.org/10.1136/jnnp.2009.195040.; Martín et al., 2001Martín, M. A., Rubio, J. C., Buchbinder, J., Fernández-Hojas, R., Hoyo, P. del y Arenas, J. et al. (2001). Molecular heterogeneity of myophosphorylase deficiency (McArdle’s disease): A genotype-phenotype correlation study. Annals of Neurology. Official Journal of the American Neurological Association and the Child Neurology Society, 50 (5), pp. 574-581.). Las pruebas diagnósticas se realizan tras una rabdomiolisis o una hiperCKemia inexplicada (X 5-10 nivel normal) (Lucía et al. 2012Lucía, A., Ruiz, J. R., Santalla, A., Nogales-Gadea, G., Rubio, J. C., García-Consuegra, I. y Navarro, C. et al. (2012). Genotypic and phenotypic features of McArdle disease: insights from the Spanish national registry. Journal of Neurology, Neurosurgery & Psychiatry, 83 (3), pp. 322-328. https://doi.org/10.1136/jnnp-2011-301593.; Martín, Lucía, Arenas y Andreu, 2014Martín, M. A., Lucía, A., Arenas, J. y Andreu, A. L. (2014). Glycogen Storage Disease Type V. GeneReviews. [En línea]. Disponible en http://www.ncbi.nlm.nih.gov/books/NBK1344/). El historial del tipo de intolerancia al ejercicio del paciente es clave para el diagnóstico. La prueba de esfuerzo del antebrazo puede realizarse de forma aérobica y demostrar un bloqueo en la glucogenolisis cuando se detecta un aumento excesivo de amonio y falta de elevación de lactato plasmático. Normalmente, la biopsia muscular se realiza en el paciente en el contexto del diagnóstico diferencial con otras miopatías o miositis; e histoquímicamente puede diagnosticarse la deficiencia de miofosforilasa. Sin embargo, actualmente el diagnostico puede realizarse a nivel genético, en muestras menos invasivas como sangre, pues la mutación “stop”, p.R50X representa en caucásicos el 60-75% de los alelos (Martín et al., 2001Martín, M. A., Rubio, J. C., Buchbinder, J., Fernández-Hojas, R., Hoyo, P. del y Arenas, J. et al. (2001). Molecular heterogeneity of myophosphorylase deficiency (McArdle’s disease): A genotype-phenotype correlation study. Annals of Neurology. Official Journal of the American Neurological Association and the Child Neurology Society, 50 (5), pp. 574-581.; Martín et al. 2014Martín, M. A., Lucía, A., Arenas, J. y Andreu, A. L. (2014). Glycogen Storage Disease Type V. GeneReviews. [En línea]. Disponible en http://www.ncbi.nlm.nih.gov/books/NBK1344/.; García-Consuegra et al., 2016García-Consuegra, I., Blázquez, A., Rubio, J. C., Arenas, J., Ballester-López, A., González-Quintana, A. y Nogales-Gadea, G. et al. (2016). Taking advantage of an old concept, “illegitimate transcription”, for a proposed novel method of genetic diagnosis of McArdle disease. Genetics in Medicine. Official Journal of the American College of Medical Genetics, 18 (11), pp. 1128-1135. https://doi.org/10.1038/gim.2015.219.). Los pacientes deben evitar contracciones musculares máximas y, lo que es más importante, practicar ejercicio moderado aeróbico a una intensidad que provoque un ritmo cardíaco inferior al 70% del máximo, para lograr un aumento en la disposición y utilización de sustratos alternativos al glucógeno e incrementar el umbral de esfuerzo necesario para provocar daño muscular. La ingesta de proteínas ayuda a activar la gluconeogénesis hepática. Una dieta adecuada de carbohidratos es necesaria para mantener la producción de glucosa a partir de glucógeno hepático y su liberación a sangre debido a que la glucosa sanguínea es un combustible alternativo clave cuando la glucogenolisis muscular está bloqueada. La ingesta de glúcidos simples previa al ejercicio físico mejora la capacidad para realizar el ejercicio (Vissing y Haller, 2003Vissing, J. y Haller, R. G. (2003). The effect of oral sucrose on exercise tolerance in patients with McArdle’s disease. The New England Journal of Medicine, 349 (26), pp. 2503-2509. https://doi.org/10.1056/NEJMoa031836.; Martín et al., 2014Martín, M. A., Lucía, A., Arenas, J. y Andreu, A. L. (2014). Glycogen Storage Disease Type V. GeneReviews. [En línea]. Disponible en http://www.ncbi.nlm.nih.gov/books/NBK1344/.).
|
Figura 1. Representación esquemática del metabolismo de los carbohidratos y las enzimas deficitarias que se asocian con patología muscular. Los enzimas afectados se encuentran sombreados (ver texto para relacionar los acrónimos) y se incluye entre paréntesis el número romano de nomenclatura correspondiente a la clasificación de las enfermedades del glucógeno
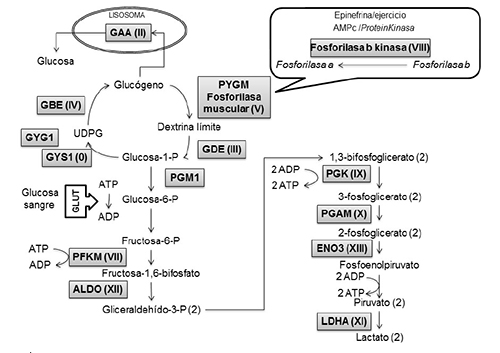
Fuente: Elaboración propia.
[Descargar tamaño completo]
| |
1.1.3. Deficiencia de fosforilasa b kinasa (GSD_IX)
La fosforilasa b quinasa (PhbK) es un homotetrámero de 4 subunidades, α, β, γ, y δ, con isoformas específicas de tejido, excepto para la subunidad β. La deficiencia de PhbK muscular es un trastorno ligado al cromosoma X, causado por mutaciones en el gen PHAK1 que codifica la isoforma muscular de la subunidad α, un componente regulador de la enzima. La PhbK fosforila al glucógeno fosforilasa, convirtiendo su forma b inactiva en la forma a activa. Por otro lado, la fosforilasa también puede activarse por AMP, IMP y fosfato inorgánico. Únicamente siete pacientes descritos (Preisler et al., 2012Preisler, N., Ørngreen, M. C., Echaniz-Laguna, A., Laforêt, P., Lonsdorfer-Wolf, E., Doutreleau, S. y Vissing, J. et al. (2012). Muscle phosphorylase kinase deficiency, a neutral metabolic variant or a disease?. Neurology, 78 (4), pp. 265-268. https://doi.org/10.1212/WNL.0b013e31824365f9.) de PhbK muscular no han mostrado alteraciones significativas del metabolismo del glucógeno durante el ejercicio. Los pacientes presentan mialgias y calambres musculares por esfuerzo, con o sin mioglobinuria, debilidad proximal o distal, o hiperCKemia asintomática (Preisler et al., 2012Preisler, N., Ørngreen, M. C., Echaniz-Laguna, A., Laforêt, P., Lonsdorfer-Wolf, E., Doutreleau, S. y Vissing, J. et al. (2012). Muscle phosphorylase kinase deficiency, a neutral metabolic variant or a disease?. Neurology, 78 (4), pp. 265-268. https://doi.org/10.1212/WNL.0b013e31824365f9.; Wehner, Clemens, Engel y Killmann, 1994Wehner, M., Clemens, P. R., Engel, A. G. y Killmann, M. W. (1994). Human muscle glycogenosis due to phosphorylase kinase deficiency associated with a nonsense mutation in the muscle isoform of the alpha subunit. Human Molecular Genetics, 3 (11), pp. 1983-1987. https://doi.org/10.1093/hmg/3.11.1983.). La biopsia muscular presenta niveles elevados de glucógeno; el diagnóstico mediante demostración de deficiencia enzimática debe confirmarse con un análisis genético.
1.1.4. Deficiencia de fosfofructoquinasa (PFK) muscular (Enfermedad de Tauri, GSD_VII)
La PFK es la enzima que cataliza el paso limitante de la glucolisis y posee tres isoformas, músculo (M), hígado (L) y plaquetas. (P). La isoforma muscular es un tetrámero de subunidades M. Los eritrocitos contienen isoformas M y L. La deficiencia de la isoforma M produce un bloqueo completo de la glucolisis muscular, y parcial en los eritrocitos. La deficiencia de PFKM impide el metabolismo oxidativo y anaeróbico, igual que en la enfermedad de McArdle. El fenotipo clínico en ambas entidades es similar; sin embargo, en la deficiencia de PFKM no se produce efecto second-wind (Haller y Vissing, 2004Haller, R. G. y Vissing, J. (2004). No spontaneous second wind in muscle phosphofructokinase deficiency. Neurology, 62 (1), pp. 82-86. https://doi.org/10.1212/WNL.62.1.82.) y la ingesta de carbohidratos es contraproducente pues inhibe la lipolisis disminuyendo los niveles séricos de ácidos grasos. Además, los pacientes presentan anemia hemolítica compensada. La enfermedad se ha descrito en diferentes fondos genéticos, pero es más prevalente en judíos Ashkenazi (Musumeci et al., 2012Musumeci, O., Bruno, C., Mongini, T., Rodolico, C., Aguennouz, M. H., Barca, E. y Toscano, A. et al. (2012). Clinical features and new molecular findings in muscle phosphofructokinase deficiency (GSD type VII). Neuromuscular Disorders, 22 (4), pp. 325-330. https://doi.org/10.1016/j.nmd.2011.10.022.). El diagnóstico histoquímico muestra ausencia de tinción PFK, aunque dada la labilidad de la enzima hay que tener en cuenta posibles errores preanalíticos. Se puede hacer diagnóstico genético confirmatorio. Los pacientes deben evitar realizar ejercicio intenso y la ingesta elevada de glúcidos previamente al ejercicio; deben también practicar ejercicio aeróbico moderado.
1.1.5. Deficiencia de fosfoglucomutasa (PGM1)
La fosfoglucomutasa 1 (PGM1) cataliza la interconversión entre glucosa-1-fosfato y glucosa-6-fosfato, siendo la isoforma mayoritaria en hígado y músculo; además, juega un papel relevante en la glicosilación normal de proteínas. La deficiencia de PGM1 muscular se ha descrito como miopatía pura, y en pacientes con defectos de la glicosilación (Tegtmeyer et al., 2014Tegtmeyer, L. C., Rust, S., Scherpenzeel, M. van, Ng, B. G., Losfeld, M. E., Timal, S. y Huijben, K. et al. (2014). Multiple phenotypes in phosphoglucomutase1 deficiency. The New England Journal of Medicine, 370 (6), pp. 533-542. https://doi.org/10.1056/NEJMoa1206605.). El uso de suplementos de galactosa parece mejorar la glicosilación y los episodios de rabdomiolisis (Preisler et al., 2013Preisler, N., Laforêt, P., Echaniz-Laguna, A., Ørngreen, M. C., Lonsdorfer-Wolf, E., Doutreleau, S. y Vissing, J. et al. (2013). Fat and carbohydrate metabolism during exercise in phosphoglucomutase type 1 deficiency. The Journal of Clinical Endocrinology & Metabolism, 98 (7), pp. E1235-E1240. https://doi.org/10.1210/jc.2013-1651.; Tegtmeyer et al., 2014Tegtmeyer, L. C., Rust, S., Scherpenzeel, M. van, Ng, B. G., Losfeld, M. E., Timal, S. y Huijben, K. et al. (2014). Multiple phenotypes in phosphoglucomutase1 deficiency. The New England Journal of Medicine, 370 (6), pp. 533-542. https://doi.org/10.1056/NEJMoa1206605.).
1.1.6. Defectos de la glucolisis distal
Los defectos de la glucolisis distal incluyen fosfoglicerato quinasa (PGK), fosfoglicerato mutasa (PGM) y lactato deshidrogenasa (LDH). Suelen ser deficiencias parciales con una elevación en los niveles de lactato de la prueba de ejercicio en el antebrazo (2X vs 4X normal). El ejercicio de máxima contracción suele inducir los síntomas. La capacidad oxidativa está conservada pues se produce suficiente piruvato para el metabolismo oxidativo dependiente de glucosa, en contraste con GSD_V o GSD_VII.
PGK (GSD IX) está codificada por un único gen, en el cromosoma X; suele producir anemia hemolítica no esferocítica que puede acompañarse por afectación del sistema nervioso central (SNC), incluyendo retardo mental, convulsiones y temblor. Un 30% presenta IE inducida por ejercicio de elevada intensidad (Spiegel et al., 2009Spiegel, R., Gómez, E. A., Akman, H. O., Krishna, S., Horovitz, Y. y DiMauro, S. (2009). Myopathic form of phosphoglycerate kinase (PGK) deficiency: a new case and pathogenic considerations. Neuromuscular Disorders, 19 (3), pp. 207-211. https://doi.org/10.1016/j.nmd.2008.12.004.).
PGAM (GSD X) es un dímero, y tiene dos isoformas, M (músculo) and B (cerebro); en músculo es un homodímero MM. Las mutaciones en la isoforma M causan miopatía pura con IE. Diez de quince casos descritos son afroamericanos con una mutación común, p.W78X. La presencia de agregados tubulares en la biopsia muscular suele ser frecuente (Naini et al., 2009Naini, A., Toscano, A., Musumeci, O., Vissing, J., Akman, H. O. y DiMauro, S. (2009). Muscle phosphoglycerate mutase deficiency revisited. Archives of Neurology, 66 (3), pp. 394-398. https://doi.org/10.1001/archneurol.2008.584.).
LDH (GSD XI) es una enzima tetramérica que contiene las subunidades muscular (LDH-A) y corazón (LDH-B), que produce un defecto puro de la glucolisis anaeróbica, conduciendo a IE con ejercicio intenso máximo y a veces dermatitis por la preponderancia de LDH-A en la piel. El diagnóstico es bioquímico y genético.
1.1.7. Otros defectos poco frecuentes
Aldolasa A (GSD_XII). Su isoforma A está presente en músculo y eritrocitos. Su deficiencia causa anemia hemolítica y miopatía (Yao et al., 2004Yao, D. C., Tolan, D. R., Murray, M. F., Harris, D. J., Darras, B. T., Geva, A. y Neufeld, E. J. (2004). Hemolytic anemia and severe rhabdomyolysis caused by compound heterozygous mutations of the gene for erythrocyte/muscle isozyme of aldolase, ALDOA(Arg303X/Cys338Tyr). Blood, 103 (6), pp. 2401-2403. https://doi.org/10.1182/blood-2003-09-3160.) inducidas por fiebre, posiblemente por la termolabilidad de la enzima.
Beta-enolasa (ENO3). Es un dímero compuesto por tres isoformas; la isoforma beta se encuentra solo en músculo; el único paciente descrito presentó IE con 5% de actividad enzimática (Comi et al., 2001Comi, G. P., Fortunato, F., Lucchiari, S., Bordoni, A., Prelle, A., Jann, S. y Bresolin, N. et al. (2001). Beta-enolase deficiency, a new metabolic myopathy of distal glycolysis. Annals of Neurology, 50 (2), pp. 202-207. https://doi.org/10.1002/ana.1095.).
Glucógeno sintasa (GYS1). Se ha descrito en cinco pacientes (tres familias) con afectación cardíaca. Algunos presentan convulsiones generalizadas. La biopsia muestra disminución de glucógeno y proliferación mitocondrial (Kollberg et al., 2007Kollberg, G., Tulinius, M., Gilljam, T., Östman-Smith, I., Forsander, G., Jotorp, P. y Holme, E. et al. (2007). Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. The New England Journal of Medicine, 357, pp. 1507-1514. https://doi.org/10.1056/NEJMoa066691.).
Glicogenina-1 (GYG1). Se ha descrito en pocos pacientes. Se produce un defecto de la autoglucosilación de la glicogenina, que es un paso clave para que comience la síntesis de glucógeno. El fenotipo es similar a GYS1 con IE, cardiomiopatía, y características similares en la biopsia muscular (Moslemi et al., 2010Moslemi, A. R., Lindberg, C., Nilsson, J., Tajsharghi, H., Andersson, B. y Oldfors, A. (2010). Glycogenin-1 deficiency and inactivated priming of glycogen synthesis. The New England Journal of Medicine, 362 (13), pp. 1203-1210. https://doi.org/10.1056/NEJMoa0900661.).
1.1.8. Alteraciones de los glúcidos que producen debilidad muscular
Deficiencia de enzima desrramificante, enfermedad de Cori, GSD_III. Es una proteína única con dos dominios, oligo-1-4-glucantransferasa y amilo-1-6-glucosidasa. Las cadenas de glucógeno son acortadas por la fosforilasa hasta restos de cuatro unidades de glucosa, que son eliminados por la desramificante en dos pasos llevados a cabo por esos dominios. Se han descrito más de 100 mutaciones. Un 85% de los casos presentan alteraciones hepatomusculares (GSD_IIIa) y un 15% únicamente hepáticas (GSD_IIIb). Los pacientes pueden desarrollar cardiomiopatía hipertrófica y arritmias (Kishnani et al., 2010Kishnani, P. S., Austin, S. L., Arn, P., Bali, D. S., Boney, A., Case, L. E. y Smit, G. P. A. et al. (2010). Glycogen storage disease type III diagnosis and management guidelines. Genetics in Medicine, 12 (7), pp. 446-463. https://doi.org/10.1097/GIM.0b013e3181e655b6.). La debilidad progresiva tiene lugar a los 30-40 años. No se produce rabdomiolisis. Es frecuente la hipertransaminemia, aumento de CK e hipoglucemia cetótica. Hay acúmulo de glucógeno anormal, que ha de confirmarse bioquímica o genéticamente. Los pacientes deben adoptar una dieta rica en proteínas con carbohidratos complejos, y deben controlarse mediante electrocardiograma y pruebas bioquímicas y ecográficas de función hepática (Kishnani et al., 2010Kishnani, P. S., Austin, S. L., Arn, P., Bali, D. S., Boney, A., Case, L. E. y Smit, G. P. A. et al. (2010). Glycogen storage disease type III diagnosis and management guidelines. Genetics in Medicine, 12 (7), pp. 446-463. https://doi.org/10.1097/GIM.0b013e3181e655b6.).
Deficiencia de enzima ramificante (GBE), enfermedad de Andersen, GSD_IV. La enzima GBE transfiere restos glucosilos unidos mediante enlace alfa-1,4 del exterior de la cadena de glucógeno a una posición alfa-1,6, sintetizando glucógeno ramificado, lo que incrementa la solubilidad y disminuye la fuerza osmótica (Thon, Khalil y Cannon, 1993Thon, V. J., Khalil, M. y Cannon, J. F. (1993). Isolation of human glycogen branching enzyme cDNAs by screening complementation in yeast. Journal of Biological Chemistry, 268 (10), pp. 7509-7513.). Hay acúmulo de glucógeno parecido a la amilopectina. Se han descrito 40 mutaciones asociadas con afectación hepática, del corazón, muscular y SNC, con formas infantiles graves y formas moderadas del adulto (Bao, Kishnani, Wu y Chen, 1996Bao, Y., Kishnani, P., Wu, J.-Y. y Chen, Y.-T. (1996). Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene. Journal of Clinical Investigation, 97 (4), pp. 941-948. https://doi.org/10.1172/JCI118517.). El diagnóstico se confirma bioquímica o genéticamente. Se debe realizar seguimiento de la afectación hepática y coronaria.
1.2. Alteraciones del metabolismo de los lípidos
En la figura 2 se muestran las vías deficitarias asociadas a miopatías lipídicas.
1.2.1. Deficiencias asociadas a debilidad muscular y acúmulo de lípidos
1.2.1.1. Deficiencia primaria de carnitina
Se produce por mutaciones en el gen OCTN2, el transportador de membrana plasmática de carnitina, que a su vez impiden la reabsorción de carnitina filtrada por el riñón. La carnitina se requiere para el transporte de ácidos grasos de cadena larga (LCFA) a la matriz mitocondrial donde tiene lugar su oxidación. Existe una presentación infantil severa con hipotonía y crisis de hipoglucemia hipocetótica, e hiperamonemia con encefalopatía hepática. En niños mayores se describe una forma que cursa con debilidad muscular, hiperCKemia y cardiomiopatía. La forma adulta solo se manifiesta con fatigabilidad (Magoulas y El-Hattab, 2012Magoulas, P. L. y El-Hattab, A. W. (2012). Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet Journal of Rare Diseases, 7 (1), 68. https://doi.org/10.1186/1750-1172-7-68.). Se producen depósitos de lípidos intramusculares, y se considera deficiencia cuando la carnitina y las acilcarnitinas en plasma y en músculo son < 10% de la normalidad, lo que puede confirmarse por recaptación impedida en linfocitos y fibroblastos. Debe seguirse la evolución de la cardiomiopatía e iniciar suplementación con L-carnitina lo antes posible.
1.2.1.2. Enfermedad de almacenamiento de lípidos neutros (NLSD)
La NLSD está causada por mutaciones en la lipasa neutra necesaria para la liberación de ácidos grasos desde los depósitos de triglicéridos en el músculo esquelético y los adipocitos. La ATGL –adipocito triglicérido lipasa–, que es activada por la proteína CGI-58, cataliza el primer paso de la hidrólisis de los triglicéridos. Las mutaciones en el gen ABHD5, que codifica CGI-58, produce NLSD con ictiocitosis (NLSDI) -síndrome de Chanarin-Dorfman- (Lefevre et al., 2001Lefevre, C., Jobard, F., Caux, F., Bouadjar, B., Karaduman, A., Heilig, R. y Özgüc, M. et al. (2001). Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. The American Journal of Human Genetics, 69 (5), pp. 1002-1012. https://doi.org/10.1086/324121.). Las mutaciones en el gen PNPLA2, que codifica ATGL (Fischer et al., 2007Fischer, J., Lefevre, C., Morava, E., Mussini, J. M., Laforêt, P., Negre-Salvayre, A. y Salvayre, R. et al. (2007). The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nature Genetics, 39 (1), pp. 28-30. https://doi.org/10.1038/ng1951.), conducen a NLSD con miopatía (NLSDM). En ambos síndromes la movilización y utilización de ácidos grasos durante el ejercicio está impedida. El fenotipo de NLSDI se caracteriza por debilidad moderada y acumulación de lípidos en músculo esquelético y en otros tejidos junto a la presencia de ictiocitosis. Los pacientes con NLSDM pueden encontrarse asintomáticos con hiperCKemia y acumulación de lípidos en músculo, corazón y otros tejidos. Estos pacientes desarrollan debilidad muscular en la tercera década de la vida, y algunos presentan cardiomiopatía dilatada. La observación de gotas lipídicas en los leucocitos de una extensión de sangre periférica -anomalía de Jordan-, junto a una intensa acumulación de lípidos en fibras musculares tipo I sugiere una posible NLSD, cuya confirmación es genética.
|
Figura 2. Esquema del metabolismo de los triglicéridos y de los ácidos grasos señalando los déficits enzimáticos que producen miopatías lipídicas. En sombreado los enzimas deficitarios (ver texto para relacionar los acrónimos)
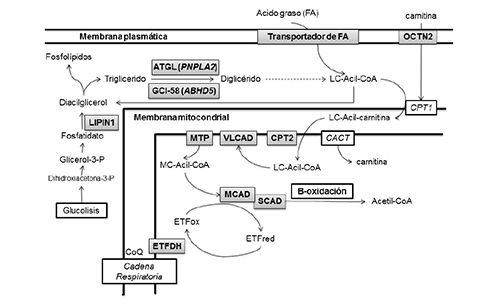
Fuente: Elaboración propia a partir de Liang, W. C y Nishino, I. (2010).
[Descargar tamaño completo]
| |
1.2.1.3. Deficiencia de mútiple acil-CoA deshidrogenasa (MADD)
MADD se produce por mutaciones en la flavoproteína transferente de electrones (ETF) A o B y por mutaciones en la deshidrogenasa (ETFDH, ETF:ubiquinona oxidorreductasa) (Laforêt y Vianey-Saban, 2010Laforêt, P. y Vianey-Saban, C. (2010). Disorders of muscle lipid metabolism: diagnostic and therapeutic challenges. Neuromuscular Disorders, 20 (11), pp. 693-700. https://doi.org/10.1016/j.nmd.2010.06.018.). La proteína transfiere electrones desde múltiples deshidrogenasas implicadas en la oxidación de ácidos grasos hasta el coenzima Q10. La clínica varía desde formas infantiles fatales hasta enfermedades multisistémicas con o sin malformaciones congénitas, pasando por enfermedades moderadas del adulto que presentan debilidad de extremidades y debilidad axial (de cuello), disfagia, IE, hepatomegalia, encefalopatía, mioglobinuria e hipoglucemia inducidas por infecciones, ayuno o cirugía (Olsen et al., 2007Olsen, R. K., Olpin, S. E., Andresen, B. S., Miedzybrodzka, Z.H., Pourfarzam, M., Merinero, B. y Andersen, O. et al. (2007). ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain, 130 (8), pp. 2045-2054. https://doi.org/10.1093/brain/awm135.), la cual es comúnmente debida a mutaciones en ETFDH. Se produce hiperCKemia, aumento de acilcarnitinas de varias longitudes y descenso de carnitina. En orina hay aciduria orgánica. En músculo hay depósitos de lípidos y algunos pacientes presentan defectos OXPHOS. El tratamiento con riboflavina mejora los síntomas en pacientes que responden con mutaciones en ETFDH. Puede observarse una deficiencia de CoQ10 secundaria tratable con suplementación.
1.2.2. Alteraciones del metabolismo lipídico con rabdomiolisis y mioglobinuria
1.2.2.1. Deficiencia de CPT2
La deficiencia de carnitina-palmitil transferasa II (CPT2) es el trastorno más común de la oxidación de los ácidos grasos y la miopatía metabólica más frecuente que cursa con rabdomiolisis recurrente y mioglobinuria (prevalencia estimada 1:300.000). La CPT (CPT1 y CPT2) está implicada en el transporte de LCFA al interior de la matriz mitocondrial, dónde se degradan por la β-oxidación. Dos formas multisistémicas, neonatal e infantil, son graves o fatales. La forma más común, miopatía del adulto, cursa con mialgias, rabdomiolisis y mioglobinuria inducidos por fiebre, ayuno o ejercicio submáximo de larga duración (Bonnefont et al., 2004Bonnefont, J.-P., Djouadi, F., Prip-Buus, C., Gobin, S., Munnich, A. y Bastin, J. (2004). Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Molecular Aspects of Medicine, 25 (5-6), pp. 495-520. https://doi.org/10.1016/j.mam.2004.06.004.). El aumento sérico de acilcarnitinas de cadena larga (C-18, 18:1, 16) hace sospechar la deficiencia, que puede confirmarse midiendo la actividad CPT II en músculo o fibroblastos, y detectando la mutación causal; en este sentido, la mutación común, p.S113L, representa el 70% de los alelos (Martín et al., 1999Martín, M. A., Rubio, J. C., Bustos, F. de, Hoyo, P. del, Campos, Y., García, A. y Arenas, J. et al. (1999). Molecular analysis in Spanish patients with muscle carnitine palmitoyltransferase deficiency. Muscle Nerve, 22 (7), pp. 941-943. https://doi.org/10.1002/(SICI)1097-4598(199907)22:7<941::AID-MUS20>3.0.CO;2-Z.). Los pacientes deben evitar el ayuno e ingerir carbohidratos cuando son físicamente activos, y ácidos grasos de cadena media (MCT). El bezafibrato aumenta los niveles de mensajero y de proteína CPT2, incrementando la oxidación LCFA. En un estudio con seis pacientes tratados con bezafibrato se mostró una mejoría en el nivel de la actividad física y del dolor muscular (Bonnefont et al., 2010Bonnefont, J.-P., Bastin, J., Laforet, P., Auby, F., Mogenet, A., Romano, S. y Djouadi, F. et al. (2010). Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clinical Pharmacology - Therapeutics, 88 (1), pp. 101-108. https://doi.org/10.1038/clpt.2010.55.).
1.2.2.2. Deficiencia de deshidrogenasa de ácidos grasos de cadena muy larga, VLCAD
La VLCAD se localiza en la membrana interna mitocondrial y cataliza el primer paso de la oxidación de LCFA. Las mutaciones stop en VLCAD producen una enfermedad infantil fatal en la infancia; sin embargo, las mutaciones con cambio de aminoácido retienen actividad residual de la enzima y producen una fenocopia de la forma adulta de CPT2. En ayuno se identifica el acúmulo sérico de acilcarnitinas de cadena larga y de la molécula C14:1 (Liang y Nishino, 2010Liang, W. C. y Nishino, I. (2010). State of the art in muscle lipid diseases. Acta Myologica, 29 (2), pp. 351-356.). El manejo del paciente es similar a la deficiencia de CPT2.
1.2.2.3. Deficiencia de Proteina Trifuncional (TP/3-hidroxiacil-CoA de cadena larga deshidrogenasa)
La TP involucra tres reacciones de la oxidación de los ácidos grasos: enoil-CoA hidratasa, 3-hydroxiacil-CoA deshidrogenasa de cadena larga (LCHAD) y acil-tiolasa, produciendo acetil-CoA y un acil-CoA acortado en dos átomos de carbono, que sirve como sustrato para un nuevo ciclo de β-oxidación. Las mutaciones afectan a todas las subunidades de la enzima, siendo la más común LCHAD, causando formas infantiles y adultas. Curiosamente algunos casos de rabdomiolisis presentan además neuropatía periférica y retinitis pigmentosa, lo que ayuda al diagnóstico diferencial con CPT2 y VLCAD. La mutación p.G510Q representa el 90% de los alelos mutantes de LCHAD. Los pacientes deben evitar el ayuno, ingerir carbohidratos y MCT (Liang y Nishino, 2010Liang, W. C. y Nishino, I. (2010). State of the art in muscle lipid diseases. Acta Myologica, 29 (2), pp. 351-356.).
1.2.2.4. Deficiencia de Lipin-1
El gen LPIN1 codifica la lipina-1, que actúa a dos niveles: como una fosfatasa ácida fosfatídica que convierte el ácido fosfatídico a diacilglicerol, el cual interviene en la síntesis de triglicéridos y fosfolípidos, y como un coactivador transcripcional que regula los genes implicados en la oxidación de ácidos grasos y en biosíntesis mitocondrial. No se conoce bien el mecanismo por el cual algunas mutaciones en este gen producen rabdomiolisis recurrente desencadenada por fiebre y ayuno, principalmente en la infancia (Michot et al., 2012Michot, C., Hubert, L., Romero, N. B., Gouda, A., Mamoune, A., Mathew, S. y Peters, H. et al. (2012). Study of LPIN1, LPIN2 and LPIN3 in rhabdomyolysis and exercise-induced myalgia. Journal of Inherited Metabolic Disease, 35 (6), pp. 1119-1128. https://doi.org/10.1007/s10545-012-9461-6.).
2. ENFERMEDADES MITOCONDRIALES Top
Las mitocondrias son orgánulos subcelulares con doble membrana que están presentes en todas las células de los mamíferos. Su principal función es la de producir la mayor parte de la energía celular en forma de ATP mediante el sistema de fosforilación oxidativa (OXPHOS), ruta final del metabolismo energético mitocondrial, que se encuentra localizado en la membrana interna de las mitocondrias. Este sistema está formado por unos 85 polipéptidos diferentes organizados en cinco complejos transmembrana (Figura 3). Los electrones procedentes de la oxidación de los carbohidratos y grasa son transportados hasta el oxígeno por medio de los cuatro primeros complejos para producir agua, al tiempo que se bombean los protones desde la matriz al espacio intermembranal. El gradiente electroquímico que se crea hace que la ATP sintasa (complejo V) pueda fosforilar el ADP produciendo el ATP. Las proteínas componentes de estos cinco complejos multienzimáticos están codificadas en el DNA mitocondrial (mtDNA) (trece polipéptidos) y el resto en el DNA nuclear nDNA (Figura 3 y Figura 4). Las mitocondrias poseen otras muchas funciones participando en la ß-oxidación, en el ciclo de la urea, en el metabolismo de aminoácidos, en la homeostasis del calcio, en la biosíntesis del grupo hemo, en la detoxificación de las especies reactivas de oxígeno y en la regulación de la apoptosis.
|
Figura 3. Esquema del sistema de fosforilación oxidativa. Subunidades codificadas en el DNA mitocondrial (azul) y en el DNA nuclear (amarillo). Puntos: coenzima Q. En la parte inferior figura el número de subunidades codificadas en uno u otro genoma
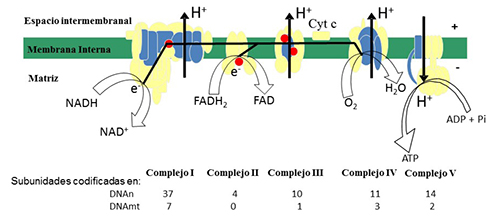
Fuente: Montoya, Emperador, López-Gallado y Ruiz-Pesini (2014).
[Descargar tamaño completo]
| |
El nombre de enfermedades mitocondriales se refiere a un amplio grupo de trastornos, causados por defectos en el sistema OXPHOS, que tienen en común el estar producidos por una deficiencia en la biosíntesis de ATP. La biogénesis del sistema OXPHOS está bajo el control de los dos sistemas genéticos de la célula (nuclear y mitocondrial), por lo que pueden mostrar distintos patrones de herencia: mendeliana (autosómica recesiva, autosómica dominante, ligada a cromosoma X) para los genes codificados en el nDNA, y materna para los codificados en el mtDNA. Las primeras mutaciones asociadas al mal funcionamiento de este sistema y que generaban enfermedades se descubrieron en el mtDNA en 1988. Desde entonces, el número de mutaciones encontradas en este genoma ha crecido enormemente, si bien alguna de ellas solo se ha presentado en un único paciente. Asimismo, se han descrito muchas mutaciones en genes de proteínas mitocondriales codificadas en el nDNA que participan en el mantenimiento y expresión del mtDNA, y en la composición y formación del sistema OXPHOS. Estas enfermedades presentan una gran variación fenotípica, con manifestaciones clínicas que afectan a distintos órganos y tejidos, por lo que su estudio requiere la participación de especialistas de muy diverso origen que aporten datos clínicos, morfológicos, bioquímicos y genéticos que permitan un diagnóstico correcto. En este trabajo nos referiremos fundamentalmente a las enfermedades debidas a alteraciones en el mtDNA y a los diversos aspectos de su diagnóstico, así como a algunos detalles de mutaciones nucleares que afectan al sistema OXPHOS, dentro de lo que se podría llamar medicina mitocondrial.
|
Figura 4. Mapa genético del DNA mitocondrial humano. RNAr, RNA ribosómico; RNAt, RNA de transferencia indicando el aminoácido que transporta; secuencias codificadoras de proteínas (ND: subunidades de la NADH deshidrogena, complejo I ; cyt b: apocitocromo b, complejo III; CO: subunidades de la citocromo c oxidasa, complejo IV, y ATP, subunidades de la ATP sintasa, complejo V
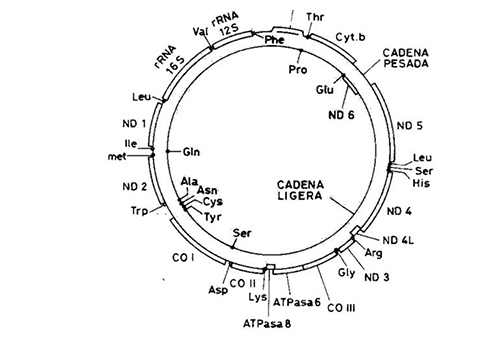
Fuente: Elaboración propia.
[Descargar tamaño completo]
| |
El mtDNA humano está formado por 16.569 pares de bases y codifica 37 genes: dos RNA ribosómicos (rRNAs), 22 RNA de transferencia (tRNAs) y 13 polipéptidos componentes de cuatro de los cinco complejos del sistema OXPHOS: siete (ND 1, 2, 3, 4, 4L, 5, 6) del complejo I o NADH:ubiquinona oxido-reductasa; uno (cyt b) del complejo III o ubiquinol: citocromo c oxido-reductasa; tres (CO I,II, III) del complejo IV o citocromo c oxidasa; y dos del complejo V o ATP sintasa (Figura 3 y Figura 4). El resto de los polipéptidos del sistema OXPHOS, así como todo el complejo II, están codificados en el nDNA. Para la síntesis de las proteínas codificadas en el mtDNA, la mitocondria utiliza un código genético que difiere del código genético universal en que el codon UGA codifica triptófano, en vez de ser un codon de terminación, los codones AUA y AUU se utilizan también como codones de iniciación, y AGA y AGG son codones de terminación en lugar de codificar para arginina. El mtDNA se hereda por vía materna. La madre trasmite el genoma mitocondrial a todos sus hijos y solamente las hijas lo pasan a todos los miembros de sucesivas generaciones. El número de moléculas de mtDNA varía entre unas pocas en las plaquetas a unas 150.000 copias en el oocito, pero la mayor parte de los tejidos contienen entre 1.000 y 10.000 por célula, con dos a diez moléculas de DNA por mitocondria (poliplasmia). Todas las moléculas de mtDNA son idénticas en tejidos normales (homoplasmia); si aparecen dos poblaciones de mtDNA, normal y mutada (heteroplasmia), estas segregan al azar entre las células hijas durante la división celular (segregación mitótica) originando tres posibles genotipos diferentes: homoplásmico para el mtDNA normal, heteroplásmico y homoplásmico para el mtDNA mutado. Por ello, el fenotipo de una célula con heteroplasmia dependerá del porcentaje de mtDNA mutado que contenga y, por tanto, de los niveles de ATP que produzca (expresión umbral) y la manifestación clínica dependerá de este nivel umbral crítico. Como consecuencia, una de las principales características de las enfermedades mitocondriales es que, en general, son multisistémicas. Los tejidos que se afectan de forma más habitual son los que requieren mayor energía (el ojo, el sistema nervioso central, el músculo esquelético, el corazón, los islotes pancreáticos, el riñón, el hígado, etc.). El mtDNA muta con una velocidad de diez a veinte veces superior a la del nDNA.
2.1. Enfermedades causadas por mutaciones en el mtDNA
Las enfermedades mitocondriales causadas por mutaciones en el mtDNA están originadas por defectos que afectan a los componentes de los complejos multienzimáticos I, III, IV y V y, por tanto, tienen en común el estar producidas por una síntesis deficiente de ATP. Las mitocondrias son componentes imprescindibles de todos los tejidos y órganos, por lo que estas enfermedades serán, en general, multisistémicas y darán lugar a un amplio espectro de fenotipos. En algunos casos, las enfermedades pueden encuadrarse en síndromes bien definidos, pero otras veces presentan solapamiento de síntomas o, como sucede en los niños, estos no se han desarrollado del todo y no están muy definidos. Asimismo, en algunos casos, las enfermedades mitocondriales pueden afectar solamente a un tejido específico, como a las células cocleares en un tipo de sordera mitocondrial o al nervio óptico en la neuropatía óptica hereditaria de Leber (LHON) (DiMauro, Hirano y Schon, 2006DiMauro, S., Hirano, M. y Schon, E. A. (2006). Mitochondrial Medicine. Abingdon, Oxon. UK: Informa Health Care. https://doi.org/10.1201/b14623.). La posibilidad de encontrarnos ante una enfermedad mitocondrial se debe tener en cuenta cuando un paciente presenta una asociación de síntomas bastante inexplicable con un rápido y progresivo curso de la enfermedad, implicando órganos no relacionados. Entre las manifestaciones clínicas más comunes se encuentran una o varias de las siguientes: encefalopatía, desórdenes motores, accidentes cerebro-vasculares, convulsiones, demencia, retraso mental, miopatía, intolerancia al ejercicio, ptosis, oftalmoplejía, retinopatía pigmentaria, atrofia óptica, ceguera, sordera, cardiomiopatía, defectos en la conducción cardiaca, disfunciones hepáticas y pancreáticas, diabetes, defectos de crecimiento, anemia sideroblástica, pseudo-obstrucción intestinal, nefropatías, acidosis metabólica y otros más secundarios. Como el número de trastornos y de mutaciones en el mtDNA es muy grande (se han descrito más de 200 mutaciones puntuales y numerosas deleciones diferentes) y son la causa de una gran variedad de enfermedades que presentan heterogeneidad en cuanto a fenotipos y a edad de aparición, en este artículo nos referiremos solamente a los fenotipos más característicos y a las mutaciones o zonas hot spot que más comúnmente se han asociado a los mismos (véase la tabla 1), dejando de lado un número muy grande de mutaciones que, en general, se han descrito solamente en casos aislados. En todo caso, habrá que tener en cuenta que nos podemos encontrar con que ciertas mutaciones que están ligadas a un fenotipo concreto puedan ser la causa de nuevos fenotipos (una misma mutación puede dar origen a síndromes muy diversos) y con que un síndrome determinado pueda estar causado por mutaciones localizadas en distintos genes. La prevalencia estimada de las enfermedades mitocondriales es de 1 cada 10.000 personas. En este trabajo clasificaremos las enfermedades de acuerdo con las características genético-moleculares de las mutaciones. Así, las enfermedades producidas por daños en el mtDNA se pueden dividir en tres grandes grupos según estén asociadas a: 1) mutaciones puntuales, 2) deleciones, 3) depleción de mtDNA.
|
Tabla 1. Mutaciones más frecuentes de DNA mitocondrial y enfermedades asociadas. Una lista actualizada de mutaciones asociadas a distintos fenotipos puede encontrarse en MITOMAP: A Human Mitochondrial Genome Database. [En línea]. Disponible en: http://www.mitomap.org
![Mutaciones más frecuentes de DNA mitocondrial y enfermedades asociadas. Una lista actualizada de mutaciones asociadas a distintos fenotipos puede encontrarse en MITOMAP: A Human Mitochondrial Genome Database. [En línea]. Disponible en: http://www.mitomap.org](https://arbor.revistas.csic.es/index.php/arbor/article/download/2274/3222/5470)
Fuente: Montoya, Emperador, López-Gallardo y Ruiz-Pesini (2014).
[Descargar tamaño completo]
| |
2.1.1. Enfermedades causadas por mutaciones puntuales en el mtDNA
El alto índice de mutación del mtDNA hace que sea posible encontrar un gran número de mutaciones puntuales; sin embargo, no todas van a ser patológicas, ya que la mayoría representan polimorfismos que se han fijado en la población y que han servido para trazar las migraciones humanas. Existen una serie de criterios para establecer cuándo se debe considerar una mutación como patogénica, pero la aplicación de los mismos depende mucho de cuán exigente se es a la hora de considerarlos. Muchas mutaciones que, en su momento, se consideraron patológicas han resultado ser simples variaciones polimórficas y, en otros casos, la secuenciación completa del mtDNA ha dado como resultado la presencia de otras mutaciones con mayor posibilidad de ser patogénicas (Montoya, López-Gallardo, Díez-Sánchez, López Pérez y Ruiz-Pesini, 2009Montoya, J., López-Gallardo, E., Díez-Sánchez, C., López Pérez, M. J. y Ruiz-Pesini, E. (2009). 20 years of human mtDNA pathologic point mutations: Carefully reading the pathogenicity criteria. Biochimica et Biophysica Acta, 1787 (5), pp. 476-483. https://doi.org/10.1016/j.bbabio.2008.09.003.; Montoya et al. 2009Montoya, J., Lopez-Gallardo, E., Herrero-Martín, M. D., Martínez-Romero, I., Gómez-Durán, A., Pacheu, D. y Ruiz-Pesini, E. et al. (2009). Diseases of the human mitochondrial oxidative phosphorylation system. Advances in Experimental Medicine and Biology, 652, pp. 47-67. https://doi.org/10.1007/978-90-481-2813-6_5.). Hasta el momento se han descrito más de 200 mutaciones puntuales patológicas distribuidas a lo largo de los 37 genes que codifica el mtDNA y que, en general, responden a un tipo de herencia materna. Algunas de estas mutaciones aparecen frecuentemente asociadas a algunos síndromes concretos (mutaciones comunes), mientras que el resto solo se han encontrado en casos puntuales. La mayor parte de las mutaciones que se han asociado a patologías determinadas, y que se consideran patogénicas, se pueden encontrar en la base de datos de MITOMAP (http://www.mitomap.org). ¿Qué mutaciones hay que estudiar ante un caso de enfermedad mitocondrial? Si los pacientes han sido evaluados con precisión, el análisis puede restringirse a unas pocas mutaciones que representan la mayor parte de los casos. Solamente ante la presencia de defectos concretos en la actividad de la cadena respiratoria se debe iniciar un estudio de la presencia de otras mutaciones descritas o nuevas. Entre las patologías más importantes causadas por mutaciones puntuales podemos citar:
2.1.1.1. Neuropatía óptica hereditaria de Leber (LHON)
Es una enfermedad específica de órgano que afecta a las células ganglionares retinales del nervio óptico y se caracteriza clínicamente por una neuropatía óptica bilateral aguda o subaguda con atrofia óptica, pérdida repentina de la visión central, edema del disco óptico, microangiopatía y un defecto grande del campo visual central. Suele aparecer entre los 20 y 40 años de edad y afecta más a hombres que a mujeres. Normalmente solo la visión está afectada, pero hay casos en los que también aparecen trastornos en la conducción cardiaca, neuropatía periférica y ataxia cerebelar. Se han descrito tres mutaciones primarias asociadas a la enfermedad: m.3460G>A, m.1778G>A y m.14484T>C, localizadas en los genes de ND1, ND4 y ND6 respectivamente. Estas mutaciones causan más del 90% de los pacientes de LHON y se heredan por vía materna. La mutación m.11778G>A es la que provoca la forma más grave de la enfermedad y es la responsable del 50% de los casos. Estas mutaciones se encuentran habitualmente en homoplasmia, pero algunos casos se han descrito en hetereoplasmia en los individuos afectados. Se han detectado otras mutaciones que se consideran patológicas, casi todas situadas en genes codificantes del complejo I (Martínez-Romero et al., 2014Martínez-Romero, I., Herrero-Martín, M. D., Llobet, L., Emperador, S., Martín-Navarro, A., Narberhaus, B. y Ruiz-Pesini, E. et al. (2014). New MT-ND1 pathologic mutation for Leber hereditary optic neuropathy. Clinical and Experimental Ophthalmology, 42 (9), pp. 856-864. https://doi.org/10.1111/ceo.12355.) y en otros genes (López-Gallardo et al., 2014López-Gallardo, E., Emperador, S., Solano, A., Llobet, L., Martín-Navarro, A., López-Pérez, M. J. y Jericó, I. et al. (2014). Expanding the clinical phenotypes of MT-ATP6 mutations. Human Molecular Genetics, 23 (23), pp. 6191-6200. https://doi.org/10.1093/hmg/ddu339.), así como combinaciones de mutaciones (Cruz-Bermúdez et al., 2016Cruz-Bermúdez, A., Vicente-Blanco, R. J., Hernández-Sierra, R., Montero, M., Álvarez, J., González Manrique, M. y Fernández-Moreno, M. A. et al. (2016). Functional Characterization of Three Concomitant mtDNA LHON Mutations Shows No Synergistic Effect on Mitochondrial Activity. PloS One, 11 (1), e0146816. https://doi.org/10.1371/journal.pone.0146816.), aunque el número de pacientes en los que se ha encontrado es muy bajo. Las mutaciones que causan LHON presentan baja penetrancia ya que la mayoría de los familiares de los pacientes son asintomáticos a pesar de poseer también la mutación en homo o heteroplasmia. Esto hace pensar en que ciertos factores ambientales o genéticos pueden ejercer una influencia en la aparición de la enfermedad.
2.1.1.2. Síndrome de NARP (Neuropatía, Ataxia y Retinopatía Pigmentaria)
Esta combinación de síntomas, a veces acompañada de retraso en el desarrollo, epilepsia, miopatía o polineuropatía, se ha descrito asociada a la mutación m.8993T>G, de herencia materna, en el gen codificante de la subunidad 6 de la ATP sintasa (complejo V). Aparece en la infancia o en adultos jóvenes. La biopsia muscular no muestra la presencia de fibras rojo-rasgadas. La mutación se encuentra en forma heteroplásmica en un rango inferior al 90% en todos los tejidos estudiados: leucocitos, fibroblastos, músculo, riñón y cerebro, aunque en nuestra casuística hemos encontrado pacientes con un porcentaje de la mutación cercano al 95%. Existe una alta correlación entre el contenido de mtDNA mutado y la severidad de la enfermedad.
2.1.1.3. Síndrome de Leigh de herencia materna (MILS)
Es una enfermedad neurodegenerativa progresiva muy grave de aparición temprana debida a una caída muy importante en la producción de energía en el cerebro en desarrollo. Las manifestaciones clínicas más evidentes son: disfunciones del tallo cerebral y de los ganglios basales, desmielinización, regresión psicomotora, retraso en el desarrollo, convulsiones, ataxia, neuropatía periférica, hipotonía, mioclonías, atrofia óptica, dificultades en la alimentación, y vómitos. Los niveles de lactato y piruvato están muy elevados en sangre y en líquido cerebro-espinal. El diagnóstico se confirma por resonancia magnética nuclear que muestra lesiones necróticas cerebrales focales en el tálamo, en el tallo cerebral y en el núcleo dentado. Esta enfermedad se produce por mutaciones en genes tanto mitocondriales como nucleares y, por tanto, se puede heredar de forma materna (mitocondrial) o mendeliana. La forma que se hereda por vía materna (MILS) está causada fundamentalmente por la mutación m.8993T>G/C, y a veces por la m.9176C>G en el gen de la subunidad 6 de la ATP sintasa del mtDNA con un alto porcentaje de heteroplasmia (por encima del 90%).
2.1.1.4. Síndrome de MELAS (encefalomiopatía mitocondrial, acidosis láctica y episodios de accidentes cerebro-vasculares)
Este síndrome está caracterizado por encefalomiopatía, acidosis láctica y accidentes cerebro-vasculares recurrentes y transitorios, producidos a edad temprana, que provocan una disfunción cerebral subaguda y cambios en la estructura cerebral con acompañamiento de hemiparesia y ceguera cortical. Además, otros caracteres comunes son: convulsiones generalizadas, migraña, sordera, demencia, vómitos, debilidad en las extremidades. La biopsia muscular suele presentar fibras rojo-rasgadas. La mayor parte de los pacientes con esta enfermedad (80%) presentan la mutación m.3243A>G localizada en el gen tRNALeu(UUR) (MT-TL1) pero también se han encontrado otras mutaciones en el mismo u otros tRNAs y alguna en genes codificantes de proteínas de los diferentes complejos. Todas las mutaciones están en forma heteroplásmica y presentan una clara herencia materna, aunque raramente más de un miembro de la familia está afectado. La mutación principal (m.3243A>G) se ha relacionado también con otras enfermedades muy distintas como oftalmoplejía progresiva externa, cardiomiopatías e incluso con diabetes mellitus y sordera, por lo que la relación genotipo-fenotipo no es muy fija. La alta frecuencia con que aparece esta mutación hace que sea de análisis obligatorio cuando estamos ante enfermedades de no muy clara sintomatología.
2.1.1.5. Síndrome de MERRF (epilepsia mioclónica con fibras rojo-rasgadas)
MERRF se caracteriza por epilepsia mioclónica acompañada a veces de miopatía, ataxia cerebelar, demencia, sordera, atrofia óptica, lipomas múltiples en cuello y tronco, con presencia de fibras rojo-rasgadas no reactivas a citocromo oxidasa. Puede aparecer tanto en la infancia como en adultos y es de curso progresivo. La mayoría de los casos de MERRF están originados por la mutación m.8344A>G localizada en el gen del MT-TK, pero también se han encontrado otras mutaciones más minoritarias en el mismo gen. Todas las mutaciones están en forma heteroplásmica.
2.1.2. Enfermedades causadas por deleciones del mtDNA
Las deleciones en el mtDNA se encuentran siempre en heteroplasmia, ya que la homoplasmia sería incompatible con la vida por faltar genes completos imprescindibles para el funcionamiento de OXPHOS. Se han descrito numerosas deleciones diferentes, si bien hay una de ellas, la llamada deleción común de 4977 pares de bases, que se encuentra mucho más frecuentemente. Los síndromes asociados a la presencia de estas deleciones son muy variados, algunos se describen a continuación.
2.1.2.1. Síndrome de oftalmoplegia externa progresiva crónica (CPEO)
Esta enfermedad es una de las más comunes entre los trastornos del mtDNA que suele aparecer en la adolescencia o en adultos y está caracterizada por oftalmoplegia, ptosis palpebral y miopatía. Además, suele ir acompañada de intolerancia al ejercicio, fatiga y debilidad de las extremidades. Las biopsias musculares presentan fibras rojo-rasgadas COX negativas. Se ha asociado fundamentalmente a deleciones grandes y únicas en mtDNA que aparecen de forma espontánea sin historia familiar. En particular en nuestros laboratorios, un 60% de pacientes con CPEO presentan la deleción común. Asimismo, esta enfermedad se ha relacionado con la mutación puntual m.3243A>G y con deleciones múltiples de herencia autosómica recesiva o dominante asociadas con mutaciones en genes como POLG, ANT1, o C10orf2.
2.1.2.2. Síndrome de Kearns-Sayre (KSS)
Este síndrome consiste en una enfermedad multisistémica progresiva caracterizada clínicamente por la presencia de CPEO, retinopatía pigmentaria y aparición antes de los 20 años de edad. Los pacientes desarrollan muy a menudo bloqueo de la conducción cardiaca, ataxia, miopatía mitocondrial, hiperproteinorraquia, sordera, demencia, fallos endocrinos y renales. La biopsia muscular muestra fibras rojo-rasgadas y COX negativas. Esta enfermedad está causada por la presencia de deleciones grandes únicas en el mtDNA de aparición espontánea, aunque también se ha asociado a deleciones múltiples (Ascaso, López-Gallardo, Prado, Ruiz-Pesini y Montoya, 2010Ascaso, F. J., López-Gallardo, E., Prado, E. del, Ruiz-Pesini, E. y Montoya, J. (2010). Macular lesion resembling adult-onset vitelliform macular dystrophy in Kearns-Sayre syndrome with multiple mtDNA deletions. Clinical and Experimental Ophthalmology, 38 (8), pp. 812-816. https://doi.org/10.1111/j.1442-9071.2010.02335.x.).
2.1.2.3. Síndrome de Pearson
El síndrome de médula ósea-páncreas de Pearson está caracterizado por anemia sideroblástica con vacuolización de precursores de la médula ósea que se manifiesta con una anemia macrocítica y disfunción pancreática exocrina que aparece en los primeros años de vida. Esta enfermedad no es esencialmente neuromuscular, pero los niños que la padecen suelen morir antes de los tres años de vida, y los pocos que sobreviven, gracias a numerosas transfusiones de sangre, suelen desarrollar posteriormente un fenotipo de Kearns-Sayre. Este síndrome está causado por deleciones grandes únicas del mtDNA de aparición esporádica.
2.1.2.4. Otras enfermedades causadas por deleciones del mtDNA
Existen otros síndromes que se han asociado a la presencia de grandes deleciones en el mtDNA. Así, en los fenotipos de diabetes, sordera y atrofia óptica, miopatías en general, DIDMOAD (diabetes mellitus, diabetes insípida, atrofia óptica y sordera). O bien a deleciones múltiples: el síndrome de MNGIE (encefalomiopatía mitocondrial neurogastrointestinal), la mioglobinuria recurrente, miositis con cuerpos de inclusión.
2.1.3. Enfermedades asociadas a depleción de mtDNA
La disminución considerable de los niveles de mtDNA es la causa de un grupo de enfermedades con caracteres clínicos y genéticos muy heterogéneos, de herencia autosómico recesiva, que aparecen muy temprano y que provocan la muerte en la infancia temprana (Lee et al., 2009Lee, N. C., Dimmock, D., Hwu, W. L., Tang, L. Y., Huang, W. C., Chinault, A. C. y Wong, L. C. (2009). Simultaneous detection of mitochondrial DNA depletion and single-exon deletion in the deoxyguanosine gene using array-based comparative genomic hybridisation. Archives of Disease in Childhood, 94 (1), pp. 55-58. https://doi.org/10.1136/adc.2008.139584.). Se presentan habitualmente con debilidad muscular, encefalomiopatía progresiva o fallo hepático. Actualmente se distinguen cuatro tipos diferentes de formas de presentación según el tipo de órgano que sufre la depleción: miopática, encefalomiopática, hepatocerebral y neurogastrointestinal (Elpeleg, 2003Elpeleg, O. (2003). Inherited Mitochondrial DNA Depletion. Pediatric Research, 54 (2), pp. 153-159. https://doi.org/10.1203/01.PDR.0000072796.25097.A5.; Suomalainen e Isohanni, 2010Suomalainen, A. e Isohanni, P. (2010). Mitochondrial DNA depletion syndromes - Many genes, common mechanisms. Neuromuscular Disorders, 20 (7), pp. 429-437. https://doi.org/10.1016/j.nmd.2010.03.017.; Nogueira et al., 2014Nogueira, C., Almeida, L. S., Nesti, C., Pezzini, I., Videira, A., Vilarinho, L. y Santorelli, F. M. (2014). Syndromes associated with mitochondrial DNA depletion. Italian Journal of Pediatrics, 40 (1), 34. https://doi.org/10.1186/1824-7288-40-34.). Se considera que existe una depleción mitocondrial cuando los niveles de mtDNA están por debajo de un 30%-20% con respecto a controles emparejados por edad y sexo (Vu et al., 1998Vu, T. H., Sciacco, M., Tanji, K., Nichter, C., Bonilla, E., Chatkupt, S. y Sharer, L. et al. (1998). Clinical manifestations of mitochondrial DNA depletion. Neurology, 50 (6), pp. 1783-1790. https://doi.org/10.1212/WNL.50.6.1783.; Morten et al., 2007Morten, K. J., Ashley, N., Wijburg, F., Hadzic, N., Parr, J., Jayawant, S. y Zeviani, M. et al. (2007). Liver mtDNA content increases during development: A comparison of methods and the importance of age- and tissue-specific controls for the diagnosis of mtDNA depletion. Mitochondrion, 7 (6), pp. 386-395. https://doi.org/10.1016/j.mito.2007.09.001.). Sin embargo, en los casos graves se llega hasta valores por debajo del 5%. La mayoría de ellos mueren en su infancia temprana, aunque algunos alcanzan hasta la pubertad e incluso mucho más (Lee et al., 2009Lee, N. C., Dimmock, D., Hwu, W. L., Tang, L. Y., Huang, W. C., Chinault, A. C. y Wong, L. C. (2009). Simultaneous detection of mitochondrial DNA depletion and single-exon deletion in the deoxyguanosine gene using array-based comparative genomic hybridisation. Archives of Disease in Childhood, 94 (1), pp. 55-58. https://doi.org/10.1136/adc.2008.139584.). Las distintas formas de presentarse la enfermedad se han asociado con mutaciones en varios genes. La forma miopática se caracteriza por una atrofia muscular proximal grave, debilidad, hipotonía, fallo en el crecimiento y dificultad para la alimentación. Esta forma se ha asociado a mutaciones en el gen timidin kinasa-2 (TK2) (Saada et al., 2001Saada, A., Shaag, A., Mandel, H., Nevo, Y., Eriksson, S. y Elpeleg, O. (2001). Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nature Genetics, 29 (3), pp. 342-344.; Mancuso et al., 2002Mancuso, M., Salviati, L., Sacconi, S., Otaegui, D., Camano, P., Marina, A. y García-Álvarez, M. et al. (2002). Mitochondrial DNA depletion. Mutations in thymidine kinase gene with myopathy and SMA. Neurology, 59 (8), pp. 1197-1202. https://doi.org/10.1212/01.WNL.0000028689.93049.9A., Cámara et al., 2015Cámara, Y., Carreño-Gago, L., Martín, M. A., Melià, M. J., Blázquez, A., Delmiro, A. y Bornstein, B. et al. (2015). Severe TK2 enzyme activity deficiency in patients with mild forms of myopathy. Neurology, 84 (22), pp. 2286-2288. https://doi.org/10.1212/WNL.0000000000001644.). La forma encefalomiopática está caracterizada por un retraso psicomotor grave, hipotonía muscular, convulsiones generalizadas, altos niveles de lactato en sangre y disfunción tubular renal variable. Se ha relacionado con mutaciones en el gen que codifica la subunidad beta de la succinil-CoA sintasa (SUCLA2) (Elpeleg et al., 2005Elpeleg, O., Miller, C., Hershkovitz, E., Bitner-Glindzicz, M., Bondi-Rubinstein, G., Rahman, S. y Saada, A. et al. (2005). Deficiency of the ADP-Forming Succinyl-CoA Synthase Activity Is Associated with Encephalomyopathy and Mitochondrial DNA Depletion. The American Journal of Human Genetics, 76 (6), pp. 1081-1086. https://doi.org/10.1086/430843.; Suomalainen e Isohanni, 2010Suomalainen, A. e Isohanni, P. (2010). Mitochondrial DNA depletion syndromes - Many genes, common mechanisms. Neuromuscular Disorders, 20 (7), pp. 429-437. https://doi.org/10.1016/j.nmd.2010.03.017.; Nogueira et al., 2014Nogueira, C., Almeida, L. S., Nesti, C., Pezzini, I., Videira, A., Vilarinho, L. y Santorelli, F. M. (2014). Syndromes associated with mitochondrial DNA depletion. Italian Journal of Pediatrics, 40 (1), 34. https://doi.org/10.1186/1824-7288-40-34.), con SUCLG1, que codifica la subunidad alfa de la succinato CoA ligasa, con la timidina fosforilasa (TYMP) y con RRMB2, ribonucleótido reductasa. Por último, la forma hepatocerebral aparece muy temprano con vómitos, fallo del desarrollo, hipotonía e hipoglucemia asociada a un deterioro neurológico progresivo. El síndrome de Alpers se considera como una forma de síndrome de depleción. El hígado muestra degeneración grasa, fibrosis, desestructuración y proliferación de conductos biliares (Mazziotta et al., 1992Mazziotta, M. R. M., Ricci, E., Bertini, E., Vici, C. D., Servidei, S., Burlina, A. B. y Moraes, C. T. et al. (1992). Fatal Infantile Liver Failure Associated with Mitochondrial DNA Depletion. The Journal of Pediatrics, 121 (6), pp. 896-901. https://doi.org/10.1016/S0022-3476(05)80335-X.). Esta forma de depleción se ha asociado a mutaciones en los genes codificantes de desoxiguanosina kinasa (DGUOK) (Mandel et al., 2001Mandel, H., Hartman, C., Berkowitz, D., Elpeleg, O. N., Manov, I. y Iancu, T. C. (2001). The hepatic mitochondrial DNA depletion syndrome: Ultrastructural changes in liver biopsies. Hepatology, 34 (4), pp. 776-784. https://doi.org/10.1053/jhep.2001.27664.), DNA polimerasa gamma (POLG) (Naviaux y Nguyen, 2004Naviaux, R. K. y Nguyen, K. V. (2004). POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Annals of Neurology, 55 (5), pp. 706-712. https://doi.org/10.1002/ana.20079.), MPV17 y TWNK (Calvo et al., 2006Calvo, S., Jain, M., Xie, X., Sheth, S. A., Chang, B., Goldberger, O. A. y Mootha, V. K. et al. (2006). Systematic identification of human mitochondrial disease genes through integrative genomics. Nature Genetics, 38 (5), pp. 576-582. https://doi.org/10.1038/ng1776.; Spinazzola et al., 2006Spinazzola, A., Viscomi, C., Fernandez-Vizarra, E., Carrara, F., D’Adamo, P., Calvo, S. y Parini, R. et al. (2006). MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nature Genetics, 38 (5), pp. 570-575. https://doi.org/10.1038/ng1765.; Nogueira et al., 2014Nogueira, C., Almeida, L. S., Nesti, C., Pezzini, I., Videira, A., Vilarinho, L. y Santorelli, F. M. (2014). Syndromes associated with mitochondrial DNA depletion. Italian Journal of Pediatrics, 40 (1), 34. https://doi.org/10.1186/1824-7288-40-34.). La primera fosforila desoxurribonucleótidos purínicos; la segunda participa en la replicación del DNA mitocondrial, y MPV17 es de función desconocida. La forma neurogastrointestinal (MNGIE) se caracteriza por una pérdida de peso, falta de movilidad gastrointestinal, disfagia, dolor abdominal, diarrea y una neuropatía desmielinizante motora y sensorial. Aparece entre los 20 y los 50 años de edad y presenta niveles elevados de timidina y desoxiuridina en plasma. Esta enfermedad puede estar producida también por deleciones múltiples y mutaciones puntuales y se ha asociado a mutaciones en el gen de la timidin fosforilasa (TYMP) y en el gen RRM2B. Mutaciones en estos genes nucleares que afectan al llamado mantenimiento del mtDNA se están asociando tanto con trastornos que producen depleción de mtDNA como con aquellos que presentan deleciones múltiples del mtDNA (Cámara et al., 2015Cámara, Y., Carreño-Gago, L., Martín, M. A., Melià, M. J., Blázquez, A., Delmiro, A. y Bornstein, B. et al. (2015). Severe TK2 enzyme activity deficiency in patients with mild forms of myopathy. Neurology, 84 (22), pp. 2286-2288. https://doi.org/10.1212/WNL.0000000000001644.).
2.2. Enfermedades causadas por mutaciones en genes nucleares codificantes de proteínas mitocondriales
Como se ha indicado anteriormente, solamente trece de las proteínas componentes del sistema OXPHOS están codificadas en el mtDNA. El resto de los polipéptidos, así como todos los factores implicados en su importe a la mitocondria, procesamiento, modificación y ensamblaje en los complejos, los enzimas y factores necesarios para el mantenimiento y expresión del genoma mitocondrial, composición fosfolipídica de la membrana interna mitocondrial y las proteínas relacionadas con la motilidad, fusión y fisión de las mitocondrias están codificados en el DNA nuclear. Por ello, cabe esperar que un gran número de enfermedades mitocondriales se deban a mutaciones en este genoma y que se presenten un modo de herencia mendeliano. La identificación de mutaciones en genes nucleares que afectan al sistema OXPHOS ha crecido muchísimo en los últimos años gracias al avance en el desarrollo de nuevas tecnologías de secuenciación de exomas y genomas completos (NGS, secuenciación de nueva generación). De este modo se ha establecido la etiología molecular de muchos fenotipos de enfermedades mitocondriales típicos o atípicos. Alguna de estas mutaciones y genes (más de 150 mitonucleares) se han descrito ya en el apartado de síndromes de depleción, y otras muchas se han descrito a lo largo de los últimos años (Taylor et al., 2014Taylor, R. W., Pyle, A., Griffin, H., Blakely, E. L., Duff, J., He, L. y Yarham, J. W. et al. (2014). Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencie. JAMA, 312 (1), pp. 68-77. https://doi.org/10.1001/jama.2014.7184.). Desde el punto de vista clínico las enfermedades causadas por mutaciones en el DNA nuclear suelen presentarse con un fenotipo más similar en los distintos pacientes de una misma familia y suelen ser más graves e incluso letales en la infancia. Una descripción de todas estas mutaciones está fuera de los objetivos de este artículo. Como hemos visto, las dificultades con las que nos encontramos en el campo de las enfermedades mitocondriales son muchísimas, lo que hace que no se tenga todavía un gran conocimiento de los mecanismos patogénicos y que no exista prácticamente ninguna estrategia terapéutica.
2.3. Tratamiento y pronóstico
No existe ninguna terapia universal para el tratamiento de las enfermedades mitocondriales. De momento, solamente pueden ser tratadas de forma paliativa dependiendo de los síntomas de cada una de ellas. Estas terapias pueden ser sintomáticas (ejercicios físicos, utilización de fármacos y cirugía), farmacológica (eliminación de metabolitos nocivos, administración de metabolitos, cofactores, administración de aceptores de electrones, eliminadores de especies reactivas de oxígeno) y genéticas (cambio de la heteroplasmia, el importe de tRNAs, y expresión alotópica de proteínas codificadas en el mtDNA). Para las enfermedades mitocondriales causadas por mutaciones en genes nucleares los mecanismos de terapia y los problemas que generan son semejantes a los de la terapia génica para otras enfermedades mendelianas.
AGRADECIMIENTOSTop
Los trabajos aquí descritos han sido subvencionados en parte por el Fondo de Investigación Sanitarias, ISCIII, MINECO (FIS-PI17-00021 – J.M.; PI17-00166; PI14-00431-M.A.M. y PI14-00903-J.A.), Diputación General de Aragón (Departamento de Ciencia, Tecnología y Universidad, Grupo de referencia B33_17R) y el Fondo Social Europeo. CIBERER es una iniciativa del Instituto de Salud Carlos III.
BIBLIOGRAFÍATop
|
| ○ | Ascaso, F. J., López-Gallardo, E., Prado, E. del, Ruiz-Pesini, E. y Montoya, J. (2010). Macular lesion resembling adult-onset vitelliform macular dystrophy in Kearns-Sayre syndrome with multiple mtDNA deletions. Clinical and Experimental Ophthalmology, 38 (8), pp. 812-816. https://doi.org/10.1111/j.1442-9071.2010.02335.x |
| ○ | Bao, Y., Kishnani, P., Wu, J.-Y. y Chen, Y.-T. (1996). Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene. Journal of Clinical Investigation, 97 (4), pp. 941-948. https://doi.org/10.1172/JCI118517 |
| ○ | Bonnefont, J.-P., Bastin, J., Laforet, P., Auby, F., Mogenet, A., Romano, S. [...] y Djouadi, F. (2010). Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clinical Pharmacology - Therapeutics, 88 (1), pp. 101-108. https://doi.org/10.1038/clpt.2010.55 |
| ○ | Bonnefont, J.-P., Djouadi, F., Prip-Buus, C., Gobin, S., Munnich, A. y Bastin, J. (2004). Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Molecular Aspects of Medicine, 25 (5-6), pp. 495-520. https://doi.org/10.1016/j.mam.2004.06.004 |
| ○ | Calvo, S., Jain, M., Xie, X., Sheth, S. A., Chang, B., Goldberger, O. A. [...] y Mootha, V. K. (2006). Systematic identification of human mitochondrial disease genes through integrative genomics. Nature Genetics, 38 (5), pp. 576-582. https://doi.org/10.1038/ng1776 |
| ○ | Cámara, Y., Carreño-Gago, L., Martín, M. A., Melià, M. J., Blázquez, A., Delmiro, A. [...] y Bornstein, B. (2015). Severe TK2 enzyme activity deficiency in patients with mild forms of myopathy. Neurology, 84 (22), pp. 2286-2288. https://doi.org/10.1212/WNL.0000000000001644 |
| ○ | Comi, G. P., Fortunato, F., Lucchiari, S., Bordoni, A., Prelle, A., Jann, S. [...] y Bresolin, N. (2001). Beta-enolase deficiency, a new metabolic myopathy of distal glycolysis. Annals of Neurology, 50 (2), pp. 202-207. https://doi.org/10.1002/ana.1095 |
| ○ | Cruz-Bermúdez, A., Vicente-Blanco, R. J., Hernández-Sierra, R., Montero, M., Álvarez, J., González Manrique, M. [...] y Fernández-Moreno, M. A. (2016). Functional Characterization of Three Concomitant mtDNA LHON Mutations Shows No Synergistic Effect on Mitochondrial Activity. PloS One, 11 (1), e0146816. https://doi.org/10.1371/journal.pone.0146816 |
| ○ | DiMauro, S., Hirano, M. y Schon, E. A. (2006). Mitochondrial Medicine. Abingdon, Oxon. UK: Informa Health Care. https://doi.org/10.1201/b14623 |
| ○ | Elpeleg, O. (2003). Inherited Mitochondrial DNA Depletion. Pediatric Research, 54 (2), pp. 153-159. https://doi.org/10.1203/01.PDR.0000072796.25097.A5 |
| ○ | Elpeleg, O., Miller, C., Hershkovitz, E., Bitner-Glindzicz, M., Bondi-Rubinstein, G., Rahman, S. [...] y Saada, A. (2005). Deficiency of the ADP-Forming Succinyl-CoA Synthase Activity Is Associated with Encephalomyopathy and Mitochondrial DNA Depletion. The American Journal of Human Genetics, 76 (6), pp. 1081-1086. https://doi.org/10.1086/430843 |
| ○ | Fischer, J., Lefevre, C., Morava, E., Mussini, J. M., Laforêt, P., Negre-Salvayre, A. [...] y Salvayre, R. (2007). The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nature Genetics, 39 (1), pp. 28-30. https://doi.org/10.1038/ng1951 |
| ○ | García-Consuegra, I., Blázquez, A., Rubio, J. C., Arenas, J., Ballester-López, A., González-Quintana, A. [...] y Nogales-Gadea, G. (2016). Taking advantage of an old concept, “illegitimate transcription”, for a proposed novel method of genetic diagnosis of McArdle disease. Genetics in Medicine. Official Journal of the American College of Medical Genetics, 18 (11), pp. 1128-1135. https://doi.org/10.1038/gim.2015.219 |
| ○ | Haller, R. G. y Vissing, J. (2002). Spontaneous second wind and glucose-induced second second wind in McArdle disease: oxidative mechanisms. Archives of Neurology, 59 (9), pp. 1395-1402. https://doi.org/10.1001/archneur.59.9.1395 |
| ○ | Haller, R. G. y Vissing, J. (2004). No spontaneous second wind in muscle phosphofructokinase deficiency. Neurology, 62 (1), pp. 82-86. https://doi.org/10.1212/WNL.62.1.82 |
| ○ | Kishnani, P. S., Austin, S. L., Arn, P., Bali, D. S., Boney, A., Case, L. E. [...] y Smit, G. P. A. (2010). Glycogen storage disease type III diagnosis and management guidelines. Genetics in Medicine, 12 (7), pp. 446-463. https://doi.org/10.1097/GIM.0b013e3181e655b6 |
| ○ | Kollberg, G., Tulinius, M., Gilljam, T., Östman-Smith, I., Forsander, G., Jotorp, P. [...] y Holme, E. (2007). Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. The New England Journal of Medicine, 357, pp. 1507-1514. https://doi.org/10.1056/NEJMoa066691 |
| ○ | Laforêt, P. y Vianey-Saban, C. (2010). Disorders of muscle lipid metabolism: diagnostic and therapeutic challenges. Neuromuscular Disorders, 20 (11), pp. 693-700. https://doi.org/10.1016/j.nmd.2010.06.018 |
| ○ | Lee, N. C., Dimmock, D., Hwu, W. L., Tang, L. Y., Huang, W. C., Chinault, A. C. y Wong, L. C. (2009). Simultaneous detection of mitochondrial DNA depletion and single-exon deletion in the deoxyguanosine gene using array-based comparative genomic hybridisation. Archives of Disease in Childhood, 94 (1), pp. 55-58. https://doi.org/10.1136/adc.2008.139584 |
| ○ | Lefevre, C., Jobard, F., Caux, F., Bouadjar, B., Karaduman, A., Heilig, R. [...] y Özgüc, M. (2001). Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. The American Journal of Human Genetics, 69 (5), pp. 1002-1012. https://doi.org/10.1086/324121 |
| ○ | Liang, W. C. y Nishino, I. (2010). State of the art in muscle lipid diseases. Acta Myologica, 29 (2), pp. 351-356. |
| ○ | Lim, J.-A., Li, L. y Raben, N. (2014). Pompe disease: from pathophysiology to therapy and back again. Frontiers in Aging Neuroscience, 6, 177. https://doi.org/10.3389/fnagi.2014.00177 |
| ○ | López-Gallardo, E., Emperador, S., Solano, A., Llobet, L., Martín-Navarro, A., López-Pérez, M. J. [...] y Jericó, I. (2014). Expanding the clinical phenotypes of MT-ATP6 mutations. Human Molecular Genetics, 23 (23), pp. 6191-6200. https://doi.org/10.1093/hmg/ddu339 |
| ○ | Lucía, A., Ruiz, J. R., Santalla, A., Nogales-Gadea, G., Rubio, J. C., García-Consuegra, I. [...] y Navarro, C. (2012). Genotypic and phenotypic features of McArdle disease: insights from the Spanish national registry. Journal of Neurology, Neurosurgery & Psychiatry, 83 (3), pp. 322-328. https://doi.org/10.1136/jnnp-2011-301593 |
| ○ | Magoulas, P. L. y El-Hattab, A. W. (2012). Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet Journal of Rare Diseases, 7 (1), 68. https://doi.org/10.1186/1750-1172-7-68 |
| ○ | Mancuso, M., Salviati, L., Sacconi, S., Otaegui, D., Camano, P., Marina, A. [...] y García-Álvarez, M. (2002). Mitochondrial DNA depletion. Mutations in thymidine kinase gene with myopathy and SMA. Neurology, 59 (8), pp. 1197-1202. https://doi.org/10.1212/01.WNL.0000028689.93049.9A |
| ○ | Mandel, H., Hartman, C., Berkowitz, D., Elpeleg, O. N., Manov, I. y Iancu, T. C. (2001). The hepatic mitochondrial DNA depletion syndrome: Ultrastructural changes in liver biopsies. Hepatology, 34 (4), pp. 776-784. https://doi.org/10.1053/jhep.2001.27664 |
| ○ | Martín, M. A., Lucía, A., Arenas, J. y Andreu, A. L. (2014). Glycogen Storage Disease Type V. GeneReviews. [En línea]. Disponible en http://www.ncbi.nlm.nih.gov/books/NBK1344/ |
| ○ | Martín, M. A., Rubio, J. C., Buchbinder, J., Fernández-Hojas, R., Hoyo, P. del [...] y Arenas, J. (2001). Molecular heterogeneity of myophosphorylase deficiency (McArdle’s disease): A genotype-phenotype correlation study. Annals of Neurology. Official Journal of the American Neurological Association and the Child Neurology Society, 50 (5), pp. 574-581. |
| ○ | Martín, M. A., Rubio, J. C., Bustos, F. de, Hoyo, P. del, Campos, Y., García, A. [...] y Arenas, J. (1999). Molecular analysis in Spanish patients with muscle carnitine palmitoyltransferase deficiency. Muscle Nerve, 22 (7), pp. 941-943. https://goo.gl/DEJTsK |
| ○ | Martínez-Romero, I., Herrero-Martín, M. D., Llobet, L., Emperador, S., Martín-Navarro, A., Narberhaus, B. [...] y Ruiz-Pesini, E. (2014). New MT-ND1 pathologic mutation for Leber hereditary optic neuropathy. Clinical and Experimental Ophthalmology, 42 (9), pp. 856-864. https://doi.org/10.1111/ceo.12355 |
| ○ | Mazziotta, M. R. M., Ricci, E., Bertini, E., Vici, C. D., Servidei, S., Burlina, A. B. [...] y Moraes, C. T. (1992). Fatal Infantile Liver Failure Associated with Mitochondrial DNA Depletion. The Journal of Pediatrics, 121 (6), pp. 896-901. https://doi.org/10.1016/S0022-3476(05)80335-X |
| ○ | Michot, C., Hubert, L., Romero, N. B., Gouda, A., Mamoune, A., Mathew, S. [...] y Peters, H. (2012). Study of LPIN1, LPIN2 and LPIN3 in rhabdomyolysis and exercise-induced myalgia. Journal of Inherited Metabolic Disease, 35 (6), pp. 1119-1128. https://doi.org/10.1007/s10545-012-9461-6 |
| ○ | Montoya, J., López-Gallardo, E., Díez-Sánchez, C., López Pérez, M. J. y Ruiz-Pesini, E. (2009). 20 years of human mtDNA pathologic point mutations: Carefully reading the pathogenicity criteria. Biochimica et Biophysica Acta, 1787 (5), pp. 476-483. https://doi.org/10.1016/j.bbabio.2008.09.003 |
| ○ | Montoya, J., Lopez-Gallardo, E., Herrero-Martín, M. D., Martínez-Romero, I., Gómez-Durán, A., Pacheu, D. [...] y Ruiz-Pesini, E. (2009). Diseases of the human mitochondrial oxidative phosphorylation system. Advances in Experimental Medicine and Biology, 652, pp. 47-67. https://doi.org/10.1007/978-90-481-2813-6_5 |
| ○ | Montoya, J., Emperador, S., López-Gallardo, E. y Ruiz-Pesini, E. (2014). Diagnóstico genético de enfermedades metabólicas producidas por alteración del DNA mitocondrial. En: Sanjurjo, P. y Baldellou, A. (eds.). Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias (4.ª ed.). Madrid: Ergon, pp. 773-790. |
| ○ | Morten, K. J., Ashley, N., Wijburg, F., Hadzic, N., Parr, J., Jayawant, S. [...] y Zeviani, M. (2007). Liver mtDNA content increases during development: A comparison of methods and the importance of age- and tissue-specific controls for the diagnosis of mtDNA depletion. Mitochondrion, 7 (6), pp. 386-395. https://doi.org/10.1016/j.mito.2007.09.001 |
| ○ | Moslemi, A. R., Lindberg, C., Nilsson, J., Tajsharghi, H., Andersson, B. y Oldfors, A. (2010). Glycogenin-1 deficiency and inactivated priming of glycogen synthesis. The New England Journal of Medicine, 362 (13), pp. 1203-1210. https://doi.org/10.1056/NEJMoa0900661 |
| ○ | Musumeci, O., Bruno, C., Mongini, T., Rodolico, C., Aguennouz, M. H., Barca, E. [...] y Toscano, A. (2012). Clinical features and new molecular findings in muscle phosphofructokinase deficiency (GSD type VII). Neuromuscular Disorders, 22 (4), pp. 325-330. https://doi.org/10.1016/j.nmd.2011.10.022 |
| ○ | Naini, A., Toscano, A., Musumeci, O., Vissing, J., Akman, H. O. y DiMauro, S. (2009). Muscle phosphoglycerate mutase deficiency revisited. Archives of Neurology, 66 (3), pp. 394-398. https://doi.org/10.1001/archneurol.2008.584 |
| ○ | Naviaux, R. K. y Nguyen, K. V. (2004). POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Annals of Neurology, 55 (5), pp. 706-712. https://doi.org/10.1002/ana.20079 |
| ○ | Nogueira, C., Almeida, L. S., Nesti, C., Pezzini, I., Videira, A., Vilarinho, L. y Santorelli, F. M. (2014). Syndromes associated with mitochondrial DNA depletion. Italian Journal of Pediatrics, 40 (1), 34. https://doi.org/10.1186/1824-7288-40-34 |
| ○ | Olsen, R. K., Olpin, S. E., Andresen, B. S., Miedzybrodzka, Z.H., Pourfarzam, M., Merinero, B. [...] y Andersen, O. (2007). ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain, 130 (8), pp. 2045-2054. https://doi.org/10.1093/brain/awm135 |
| ○ | Preisler, N., Laforêt, P., Echaniz-Laguna, A., Ørngreen, M. C., Lonsdorfer-Wolf, E., Doutreleau, S. [...] y Vissing, J. (2013). Fat and carbohydrate metabolism during exercise in phosphoglucomutase type 1 deficiency. The Journal of Clinical Endocrinology & Metabolism, 98 (7), pp. E1235-E1240. https://doi.org/10.1210/jc.2013-1651 |
| ○ | Preisler, N., Ørngreen, M. C., Echaniz-Laguna, A., Laforêt, P., Lonsdorfer-Wolf, E., Doutreleau, S. [...] y Vissing, J. (2012). Muscle phosphorylase kinase deficiency, a neutral metabolic variant or a disease?. Neurology, 78 (4), pp. 265-268. https://doi.org/10.1212/WNL.0b013e31824365f9 |
| ○ | Quinlivan, R., Buckley, J., James, M., Twist, A., Ball, S., Duno, M. [...] y Winer, J. (2010). McArdle disease: a clinical review. Journal of Neurology, Neurosurgery & Psychiatry, 81 (11), pp. 1182-1188. https://doi.org/10.1136/jnnp.2009.195040 |
| ○ | Saada, A., Shaag, A., Mandel, H., Nevo, Y., Eriksson, S. y Elpeleg, O. (2001). Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nature Genetics, 29 (3), pp. 342-344. |
| ○ | Sharp, L. J. y Haller, R. G. (2014). Metabolic and mitochondrial myopathies. Neurologic Clinics, 32 (3), pp. 777-799. https://doi.org/10.1016/j.ncl.2014.05.001 |
| ○ | Spiegel, R., Gómez, E. A., Akman, H. O., Krishna, S., Horovitz, Y. y DiMauro, S. (2009). Myopathic form of phosphoglycerate kinase (PGK) deficiency: a new case and pathogenic considerations. Neuromuscular Disorders, 19 (3), pp. 207-211. https://doi.org/10.1016/j.nmd.2008.12.004 |
| ○ | Spinazzola, A., Viscomi, C., Fernandez-Vizarra, E., Carrara, F., D’Adamo, P., Calvo, S.[...] y Parini, R. (2006). MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nature Genetics, 38 (5), pp. 570-575. https://doi.org/10.1038/ng1765 |
| ○ | Suomalainen, A. e Isohanni, P. (2010). Mitochondrial DNA depletion syndromes - Many genes, common mechanisms. Neuromuscular Disorders, 20 (7), pp. 429-437. https://doi.org/10.1016/j.nmd.2010.03.017 |
| ○ | Taylor, R. W., Pyle, A., Griffin, H., Blakely, E. L., Duff, J., He, L. [...] y Yarham, J. W. (2014). Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencie. JAMA, 312 (1), pp. 68-77. https://doi.org/10.1001/jama.2014.7184 |
| ○ | Tegtmeyer, L. C., Rust, S., Scherpenzeel, M. van, Ng, B. G., Losfeld, M. E., Timal, S. [...] y Huijben, K. (2014). Multiple phenotypes in phosphoglucomutase1 deficiency. The New England Journal of Medicine, 370 (6), pp. 533-542. https://doi.org/10.1056/NEJMoa1206605 |
| ○ | Thon, V. J., Khalil, M. y Cannon, J. F. (1993). Isolation of human glycogen branching enzyme cDNAs by screening complementation in yeast. Journal of Biological Chemistry, 268 (10), pp. 7509-7513. |
| ○ | Vissing, J. y Haller, R. G. (2003). The effect of oral sucrose on exercise tolerance in patients with McArdle’s disease. The New England Journal of Medicine, 349 (26), pp. 2503-2509. https://doi.org/10.1056/NEJMoa031836 |
| ○ | Vu, T. H., Sciacco, M., Tanji, K., Nichter, C., Bonilla, E., Chatkupt, S. [...] y Sharer, L. (1998). Clinical manifestations of mitochondrial DNA depletion. Neurology, 50 (6), pp. 1783-1790. https://doi.org/10.1212/WNL.50.6.1783 |
| ○ | Wehner, M., Clemens, P. R., Engel, A. G. y Killmann, M. W. (1994). Human muscle glycogenosis due to phosphorylase kinase deficiency associated with a nonsense mutation in the muscle isoform of the alpha subunit. Human Molecular Genetics, 3 (11), pp. 1983-1987. https://doi.org/10.1093/hmg/3.11.1983 |
| ○ | Yao, D. C., Tolan, D. R., Murray, M. F., Harris, D. J., Darras, B. T., Geva, A. y Neufeld, E. J. (2004). Hemolytic anemia and severe rhabdomyolysis caused by compound heterozygous mutations of the gene for erythrocyte/muscle isozyme of aldolase, ALDOA(Arg303X/Cys338Tyr). Blood, 103 (6), pp. 2401-2403. https://doi.org/10.1182/blood-2003-09-3160 |
|