MEDICINA, CIENCIA Y REALIDAD DE LAS ENFERMEDADES RARAS / MEDICINE, SCIENCE AND REALITY OF RARE DISEASES
CUANDO EL CÁNCER ES UNA ENFERMEDAD RARA
Miguel Urioste
Centro Nacional de Investigaciones Oncológicas (CNIO) / Centro de Investigaciones Biomédicas en Red de Enfermedades Raras (CIBERER)
ORCID iD: http://orcid.org/0000-0001-7583-7305
murioste@cnio.es
Javier Benítez
Centro Nacional de Investigaciones Oncológicas (CNIO) / Centro de Investigaciones Biomédicas en Red de Enfermedades Raras (CIBERER)
ORCID iD: http://orcid.org/0000-0002-0923-7202
jbenitez@cnio.es
RESUMEN
Heredar una alteración en un gen de susceptibilidad al cáncer supone vivir con una probabilidad alta de desarrollar la enfermedad, con frecuencia más de una vez, y por lo general a una edad temprana. Y también convivir con otros familiares, padres, hijos, o hermanos, que pasan por idénticas circunstancias. Existen 200 entidades clínicas diferentes en las que se hereda la susceptibilidad al cáncer y todas ellas tienen un enorme impacto personal y familiar. Aunque en su mayoría son poco frecuentes, en su conjunto representan una parte sustancial, en torno al 5%, del conjunto de cánceres, por lo que también su impacto poblacional resulta importante. La identificación de estas personas o familias y su derivación a unidades especializadas para que reciban un adecuado asesoramiento genético y posterior seguimiento clínico contribuye a aliviar a las familias, a la vez que evita costes sanitarios innecesarios restringiendo las medidas de seguimiento solo a aquellos que las necesitan. En este proceso el conocimiento de los genes responsables y el estudio genético de las familias en riesgo es un paso de importancia crucial. Las nuevas técnicas de secuenciación masiva del exoma han facilitado la búsqueda de nuevos genes responsables del cáncer familiar y de síndromes de susceptibilidad al cáncer que a corto plazo proporcionarán un espectro más correcto y completo de los mismos y a medio plazo permitirán una aplicación masiva en la práctica clínica.
WHEN CANCER IS A RARE DISEASE
ABSTRACT
When a person inherits a mutation in a cancer susceptibility gene it means that he/she will have a high probability of developing cancer at an early age of onset and will live with several first and second degree familial members in the same situation. There are more than 200 clinical entities with cancer susceptibility and all of them have a familial and personal impact. Although their frequency is low, corresponding to around 5% of cancers, they have a high impact on the population. The identification of people at risk and their clinical follow up are important to provide adequate genetic counseling and help to reduce costs by focusing the different surveillance measures only on people with the highest probability of developing cancer. Determining the genes responsible and conducting a genetic study in families at risk constitute critical steps in this process. New technologies of massive sequencing have facilitated the search for genes associated with familial cancer. We expect to soon have a wide spectrum of the genetic bases of these syndromes and to be able to apply these results to clinical practice..
Recibido: 30-04-2015; Aceptado: 13-05-2016.
Cómo citar este artículo/Citation: Urioste, M. y Benítez, J. (2018). Cuando el cáncer es una enfermedad rara. Arbor, 194 (789): a464. https://doi.org/10.3989/arbor.2018.789n3006
PALABRAS CLAVE: Cáncer familiar; cáncer hereditario; síndrome de susceptibilidad al cáncer; genes de alta susceptibilidad; secuenciación masiva del exoma; asesoramiento genético.
KEYWORDS: Familial cancer; hereditary cancer; cancer susceptibility syndromes; massive exome sequencing; genetic counseling.
Copyright: © 2018 CSIC. Este es un artículo de acceso abierto distribuido bajo los términos de la licencia de uso y distribución Creative Commons Reconocimiento 4.0 Internacional (CC BY 4.0).
CONTENIDOS
INTRODUCCIÓN Top
El término cáncer engloba un grupo numeroso de enfermedades que se caracterizan por el desarrollo de células anormales, que se dividen y crecen sin control en cualquier parte del cuerpo. Durante 2018, 1.700.000 personas serán diagnosticadas de cáncer en los Estados Unidos y 609.640 personas morirán por esta enfermedad (American Cancer Society, 2018American Cancer Society (2018). Cancer Facts & Figures 2018. Atlanta, Ga: American Cancer Society.). En 2017 en España se esperan 228.482 nuevos casos de cáncer, y 315.413 para el año 2035 (Sociedad Española de Oncología Médica, SEOM, 2018Sociedad Española de Oncología Médica (SEOM). Las cifras del cáncer en España 2018. [En línea]. Disponible en https://www.seom.org/seomcms/images/stories/recursos/Las_Cifras_del_cancer_en_Espana2018.pdf.). Si bien las cifras de incidencia y mortalidad ayudan a comprender la magnitud de la carga que representa esta enfermedad para la sociedad, estos indicadores contribuyen poco a entender el efecto del cáncer sobre los pacientes y sus familias. Además de la morbilidad física que causa el cáncer, esta enfermedad conlleva un fuerte sufrimiento emocional y una reducción general de la calidad de vida del paciente y de sus familiares (Faller et al., 2013Faller, H., Schuler, M., Richard, M., Heckl, U., Weis, J. y Küffner, R. (2013). Effects of psycho-oncologic interventions on emotional distress and quality of life in adult patients with cancer: systematic review and meta-analysis. Journal of Clinical Oncology, 31 (6), pp. 782-793. https://doi.org/10.1200/JCO.2011.40.8922.).
La mayor parte de los cánceres tiene un carácter esporádico, es decir, aparecen en individuos en los que no existe ningún riesgo familiar o hereditario de desarrollar la enfermedad. En estos casos la enfermedad aparece por lo general a edades avanzadas, generalmente más allá de los 65 años, y es consecuencia de la acumulación a lo largo de esos años de alteraciones genéticas en las células de un determinado tejido. Sin embargo, existe un pequeño porcentaje de casos, entre un 3 y un 10% del total de cánceres, en los que el desarrollo del cáncer está relacionado con la presencia de un síndrome de predisposición genética al cáncer (SPGC). Son los casos que suelen denominarse cáncer hereditario y que se identifican en base a ciertas características personales y familiares.
Los casos hereditarios se deben a factores genéticos de susceptibilidad que porta el individuo desde su nacimiento, factores que hereda de sus progenitores y que transmitirá a sus descendientes. Es importante insistir en que no es el cáncer lo que se hereda, sino la predisposición o susceptibilidad genética a desarrollarlo, circunstancia que no siempre implica una probabilidad del 100% de tener la enfermedad, sino que el riesgo de desarrollarla es significativamente superior al observado en la población general. La identificación de individuos y familias con un riesgo incrementado de desarrollar cáncer permite, además de una valoración individualizada del riesgo de desarrollar la enfermedad, recomendar estrategias de prevención y diagnóstico precoz adecuadas en cada caso.
Hasta la fecha se han descrito alrededor de 200 SPGC, la mayoría de ellos son poco frecuentes, y en todos ellos el riesgo de cáncer excede el riesgo poblacional, si bien las cifras de riesgo son muy diferentes de unos a otros. Además, estos síndromes manifiestan una gran variación en su expresividad, de modo que, dentro de una misma familia podemos observar marcadas diferencias en la edad de aparición, el tipo de tumor, su localización, en la agresividad o en la tasa de supervivencia.
La práctica totalidad de los SPGC conocidos hasta la fecha son procesos monogénicos y, en consecuencia, se heredan siguiendo patrones mendelianos. Sin una adecuada historia familiar, muchos SPGC podrían no identificarse y ser considerados como tumores de carácter esporádico. La historia familiar es sin duda la herramienta más eficaz para determinar la probabilidad de que una familia tenga un SPGC, y para poder llevar a cabo el proceso de asesoramiento genético (Bennett et al., 1995Bennett, R. L., Steinhaus, K. A., Uhrich, S. B., O’Sullivan, C. K., Resta, R. G., Lochner-Doyle, D. [...] y Hamanishi, J. (1995). Recommendations for standardized human pedigree nomenclature. The American Journal of Human Genetics, 56, pp. 745-752.).
EL CÁNCER HEREDITARIO COMO ENFERMEDAD RARA Top
Las cifras mencionadas al inicio demuestran que el cáncer en su conjunto es una enfermedad muy común, si bien algunas formas de cáncer y en particular las formas hereditarias y los SPGC entran dentro del concepto de enfermedades raras. En la tabla 1 se muestran los SPGC más comunes, en los que se conoce el gen o los genes responsables. Además del nombre de los genes, la tabla también recoge el número con el que la entidad se reconoce en el catálogo de McKusick (1998McKusick, V. A. (1998). Mendelian Inheritance in Man: a catalog of human genes and genetic disorders. Johns Hopkins University.), las cifras de penetrancia y de riesgo de cáncer, y la incidencia del síndrome en la población.
|
Tabla 1. Síndromes de predisposición al cáncer más frecuentes
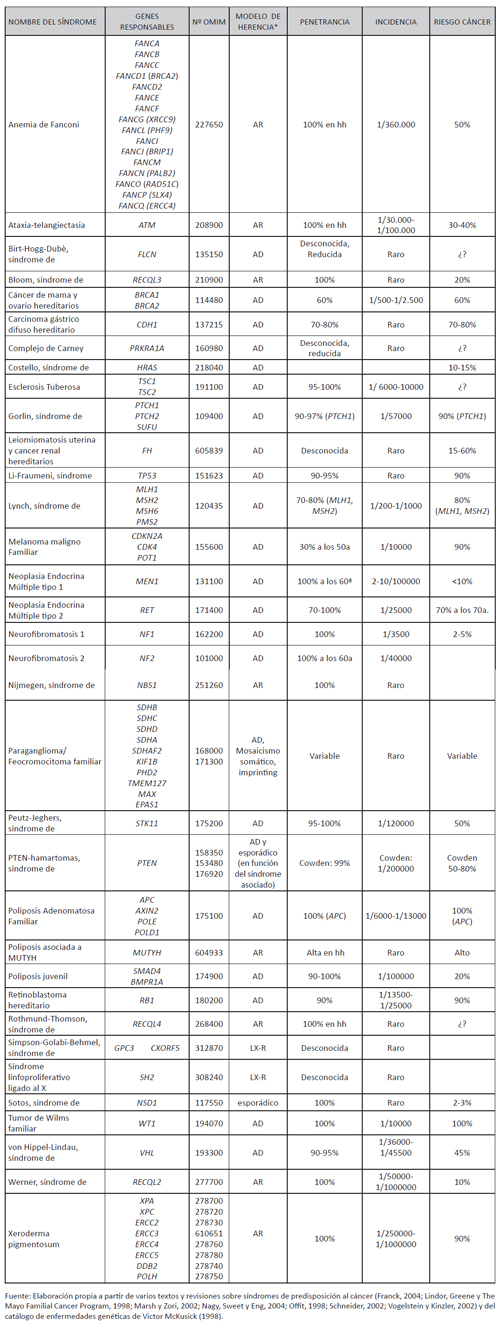
Fuente: Elaboración propia a partir de varios textos y revisiones sobre síndromes de predisposición al cáncer (Frank, 2004; Lindor, Greene y The Mayo Familial Cancer Program, 1998; Marsh y Zori, 2002; Nagy, Sweet y Eng, 2004; Offit, 1998; Schneider, 2002; Vogelstein y Kinzler, 2002) y del catálogo de enfermedades genéticas de Victor McKusick (1998).
[Descargar tamaño completo]
| |
Con la excepción del Síndrome de Cáncer de Mama y Ovario Hereditario (SCMOH) y del Síndrome de Lynch, todas las demás entidades se ajustan a la definición de enfermedad rara de la Comunidad Europea. Todos muestran frecuencias inferiores a los cinco casos por 10.000 habitantes. Presentan además grandes dificultades diagnósticas y de seguimiento, conllevan serios problemas sanitarios, un fuerte impacto personal y familiar, de la mayoría existe escasa información epidemiológica, y plantean importantes dificultades para su investigación por su escasa frecuencia y por la alta mortalidad que conlleva la enfermedad. Además, en muchos de los SPGC clínicamente definidos se desconoce la causa o causas genéticas. Por ejemplo, en las formas familiares de cáncer de testículo, de próstata, de células escamosas de esófago, de carcinoide gástrico, el esófago de Barret, el cáncer colorrectal familiar de tipo X, en algunos tumores cerebrales familiares, etc. A este grupo de SPGC de causa desconocida hay que añadir todas aquellas familias en las que no se identifica la mutación responsable aun estando diagnosticadas de un síndrome con gen conocido. Así, en el 40% de las familias que se ajustan a los criterios clínicos del SCMOH, no se detecta la mutación causal ni en BRCA1 y BRCA2, ni en otros genes asociados al desarrollo de la enfermedad (PALB2, BRIP1, RAD51C, TP53, PTEN, etc.) (Southey et al., 2013Southey, M. C., Park, D. J., Nguyen-Dumont, T., Campbell, I., Thompson, E., Trainer, A. H. [...] y Goldgar, D. E. (2013). COMPLEXO: identifying the missing heritability of breast cancer via next generation collaboration. Breast Cancer Ressearch, 15 (3), pp. 402-402. https://doi.org/10.1186/bcr3434.). Igualmente, en el 40-50% de los casos de cáncer colorrectal no-polipósico hereditario (HNPCC) que se ajustan a los criterios clínicos más exigentes (Amsterdam I), no se identifican mutaciones en ninguno de los genes de reparación de bases desapareadas (MMR, mismatch repair genes) (Valle et al., 2007Valle, L., Perea, J., Carbonell, P., Fernández, V., Dotor, A. M., Benítez, J. y Urioste, M. (2007). Clinicopathologic and pedigree differences in Amsterdam I-positive hereditary nonpolyposis colorrectal cancer families according to tumor microsatellite instability status. Journal of Clinical Oncology, 25 (7), pp. 781-786. https://doi.org/10.1200/JCO.2006.06.9781.). También en el 10% de las familias con formas clásicas de poliposis adenomatosa familiar (PAF) y en el 60-70% de las formas atenuadas de PAF no se detectan mutaciones en los genes conocidos asociados a poliposis adenomatosa (APC, MUTYH, AXIN2, POLD1 y POLE) (Rivera et al., 2014Rivera, B., Perea, J., Sánchez, E., Villapun, M., Sánchez-Tomé, E., Mercadillo, F. [...] y Urioste, M. (2014). A novel AXIN2 germline variant associated with attenuated FAP without signs of oligondontia or ectodermal dysplasia. European Journal of Human Genetics, 22 (3), pp. 423-426. https://doi.org/10.1038/ejhg.2013.146.). Aunque en distintas proporciones esta situación se da en todos los SPGC conocidos. La heterogeneidad genética es por tanto la norma en los SPGC y una parte sustancial de la misma es desconocida.
IDENTIFICACIÓN DE LOS SÍNDROMES DE PREDISPOSICIÓN AL CÁNCER Top
Más del 90% de los cánceres son esporádicos, es decir, los factores ambientales tienen un peso importante en su aparición. Existen numerosos factores ambientales potencialmente cancerígenos, muchos de ellos están recogidos en el capítulo primero de libro de K. Schneider (Schneider, 2002Schneider, K. (2002). Counselling About Cancer. Strategies for Genetic Counselling (2.ª ed.). New York: Wiley.). Por el contrario, solo un 3-10% de todos los tumores muestran un claro carácter hereditario y en su aparición el papel principal es de los factores genéticos frente a los ambientales. Entre todos los cánceres es importante poder identificar estos casos hereditarios, ya que mediante el consejo genético los individuos y las familias afectadas van a poder beneficiarse de las medidas de prevención y detección precoz que conllevan una efectiva reducción de la mortalidad.
El cáncer hereditario presenta una serie de características clínicas cuyo conocimiento favorece su identificación. Las principales características son (Urioste Azcorra, 2011Urioste Azcorra, M. (2011). Detección e identificación de síndromes de susceptibilidad al cáncer. En: Bandrés Moya, F. y Urioste Azcorra, M. (eds.). Planteamientos básicos del cáncer hereditario: principales síndromes. Madrid: Fundación Tejerina, pp. 27-46.):
Alta incidencia de cáncer en la familia, que suele ser la señal de alarma más común y la principal causa de consulta.
Aparición del mismo tipo de cáncer, que por lo general se presenta en generaciones sucesivas, o en una única generación, siguiendo modelos mendelianos.
Aparición del cáncer a edad temprana, entre 10-15 años antes de la edad en la que es frecuente la aparición de la forma esporádica de ese mismo tipo de tumor.
Bilateralidad en el caso de afectación de órganos pares.
Multifocalidad, iniciándose el tumor de manera independiente en varios focos repartidos por el órgano donde se asienta.
Aparición de varios cánceres primarios en el mismo individuo. Al valorar esta circunstancia conviene tener presente que las segundas neoplasias en ocasiones pueden guardar relación con el tratamiento de neoplasias anteriores.
Asociación del cáncer con defectos del desarrollo, dando lugar a fenotipos complejos donde el cáncer es un rasgo más dentro de un conjunto de manifestaciones o signos, en el que son comunes defectos del desarrollo mayores y menores.
La identificación de una o varias de estas características debe alertar sobre la existencia de un posible caso de cáncer hereditario y debe iniciarse una evaluación especializada que confirme la sospecha diagnóstica y lleve a cabo la realización de pruebas genéticas específicas, el consejo genético del individuo y su familia y establezca las oportunas medidas de seguimiento, detección precoz y reducción del riesgo.
SÍNDROME DE PTEN-TUMORES HAMARTOMATOSOS Top
Aunque el 70% de las consultas de cáncer hereditario giran en torno al cáncer de mama/ovario y las formas polipósicas y no polipósicas de cáncer colorrectal, otros SPGC, como el Síndrome de PTEN-tumores hamartomatosos, (SPTH), aunque menos comunes en la práctica diaria, ilustran mejor la dificultad que entraña el reconocimiento y el manejo de estos pacientes. El SPTH es un fiel reflejo de los problemas que plantea cualquier enfermedad rara, y a continuación revisaremos el estado actual del conocimiento del síndrome tratando de mostrar la complejidad y las dificultades para avanzar en la definición del fenotipo, de sus bases moleculares y de las mejores opciones para el manejo y tratamiento de los pacientes.
El término SPTH engloba un conjunto de entidades clínicas causadas por mutaciones germinales en el gen PTEN, entre las que se encuentran los síndromes de Cowden (SC), y de Bannayan-Riley-Ruvalcaba (SBRR) -los dos principales-, la enfermedad de Lhermitte-Duclos (ELD), los trastornos del espectro autista asociados a macrocefalia y la entidad conocida como Proteus-like. El SC es el prototipo dentro del SPTH. Es un síndrome raro, difícil de reconocer -en parte porque una alta proporción de individuos de la población general presentan uno o más rasgos del síndrome-, multisistémico, caracterizado por la presencia de tumores de carácter benigno junto a la predisposición para desarrollar cáncer, descrito clínicamente hace más de 30 años antes de identificarse las mutaciones germinales en el gen PTEN como causa del mismo (Lloyd y Dennis, 1963Lloyd, K. M. y Dennis, M. (1963). Cowden´s disease: a possible new symptom complex with multiple system involvement. Annals of Internal Medicine, 58 (1), pp. 136-142. https://doi.org/10.7326/0003-4819-58-1-136.). Por su parte el SBRR es una entidad aún más rara, que se manifiesta en el nacimiento, caracterizada por macrocefalia, sobrecrecimiento, hamartomas, lipomatosis, alteraciones vasculares, retraso psicomotor y máculas hiperpigmentadas en el pene (Gorlin, Cohen, Condon y Burke, 1992Gorlin, R. J., Cohen Jr., M. M., Condon, L. M. y Burke, B. A. (1992). Bannayan-Riley-Ruvalcaba syndrome. American Journal of Medical Genetics, 44 (3), pp. 307-314. https://doi.org/10.1002/ajmg.1320440309.).
Fenotipo y criterios diagnósticos
Al tratarse de una enfermedad rara, con una alta variación fenotípica, en 1983 se propusieron los primeros criterios diagnósticos para el SC (Salem y Steck, 1983Salem, O. S. y Steck, W. D. (1983). Cowden´s disease (multiple hamartoma and neoplasia syndrome): A case report and review of the English literature. Journal of the American Academy of Dermatology, 8 (5), pp. 686-696. https://doi.org/10.1016/S0190-9622(83)70081-2), que fueron revisados por un consorcio internacional de expertos en 1996 (Nelen et al., 1996Nelen, M. R., Padberg, G. W., Peeters, E. A. J., Lin, A. Y., Helm, B. van den, Frants, R. R. [...] y Eng, C. (1996). Localization of the gene for Cowden disease to chromosome 10q22-23. Nature Genetics, 13 (1), pp. 114-116. https://doi.org/10.1038/ng0596-114.). A partir de esta fecha el diagnóstico del síndrome se ha fundamentado en las distintas versiones de los criterios del consorcio. La tabla 2 recoge los criterios del consorcio publicados en el 2000. Sin lugar a dudas estos criterios han sido útiles para la identificación del síndrome y para el manejo de pacientes y familias. No obstante, los trabajos y revisiones recientes insisten en las dificultades diagnósticas y han ampliado el espectro de alteraciones asociadas al síndrome, modificando el valor de algunas de ellas en el proceso diagnóstico (los criterios revisados aparecen en la tabla 3) (Tan et al., 2011Tan, M. H., Mester, J., Peterson, C., Yang, Y., Chen, J. L., Rybicki, L. A [...] y Eng, C. (2011). A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. The American Journal of Human Genetics, 88 (1), pp. 42-56. https://doi.org/10.1016/j.ajhg.2010.11.013.; Pilarski et al., 2013Pilarski, R., Burt, R., Kohlman, W., Pho, L., Shannon, K. M. y Swisher, E. (2013). Cowden Syndrome and the PTEN Hamartoma Tumor Syndrome: Systematic review and revised diagnostic criteria. Journal of the National Cancer Institute, 105 (21), pp. 1607-1616. https://doi.org/10.1093/jnci/djt277.).
|
Tabla 2. Criterios diagnósticos del síndrome de Cowden

Fuente: Elaboración propia a partir de International Cowden Syndrome Consortium, 2000 (Eng, 2001).
[Descargar tamaño completo]
| |
|
Tabla 3. Criterios diagnósticos revisados del síndrome de PTEN-tumores hamartomasos

Fuente: Elaboración propia a partir de Tan et al., 2011 y Pilarski, Burt, Kohlman, Pho, Shannon y Swisher, 2013.
[Descargar tamaño completo]
| |
A la dificultad diagnóstica contribuye la aparición dependiente de la edad de ciertas manifestaciones, circunstancia por la que la identificación del síndrome está condicionada por el servicio de salud que valore al paciente. Así, los pediatras están muy habituados a medir el perímetro cefálico de sus pacientes y pueden sospechar el síndrome ante una macrocefalia. No sucede los mismo con los gastroenterólogos u oncólogos, para los que la presencia de pólipos o de cáncer, serán determinantes a la hora de sospechar el síndrome. De hecho, la presencia de alguno de estos dos signos suele ser uno de los motivos más frecuentes para indicar el estudio de PTEN en un adulto. Por otro lado, la identificación de alteraciones mucocutáneas propias del síndrome, que históricamente han tenido un gran valor de cara al diagnóstico, requiere una formación muy específica de la que carecen la mayor parte de los profesionales médicos (Tan et al., 2011Tan, M. H., Mester, J., Peterson, C., Yang, Y., Chen, J. L., Rybicki, L. A [...] y Eng, C. (2011). A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. The American Journal of Human Genetics, 88 (1), pp. 42-56. https://doi.org/10.1016/j.ajhg.2010.11.013.). La extensa revisión de la bibliografía llevada a cabo por Pilarski et al. (2013Pilarski, R., Burt, R., Kohlman, W., Pho, L., Shannon, K. M. y Swisher, E. (2013). Cowden Syndrome and the PTEN Hamartoma Tumor Syndrome: Systematic review and revised diagnostic criteria. Journal of the National Cancer Institute, 105 (21), pp. 1607-1616. https://doi.org/10.1093/jnci/djt277.) ha revelado que: 1) no hay suficientes evidencias para considerar como patognomónica ninguna de las características descritas en el SC; 2) tampoco hay evidencias para considerar la mastopatía fibroquística mamaria, ni los miomas uterinos, ni las malformaciones genitourinarias, ni los tumores del SNC -con excepción del ELD- como criterios diagnósticos; 3) en cambio, sí deberían ser incluidos como criterios diagnósticos los trastornos del espectro autista, el cáncer colorrectal, la acantosis glucogénica del esófago, las máculas en el glande del pene, el carcinoma de células renales, la lipomatosis testicular y las anomalías vasculares; y 4) la información reciente sugiere que los criterios del consorcio internacional utilizados hasta la fecha son demasiado laxos y deben ser sustituidos por criterios más restrictivos, como los que proponen estos autores (Pilarski et al., 2013Pilarski, R., Burt, R., Kohlman, W., Pho, L., Shannon, K. M. y Swisher, E. (2013). Cowden Syndrome and the PTEN Hamartoma Tumor Syndrome: Systematic review and revised diagnostic criteria. Journal of the National Cancer Institute, 105 (21), pp. 1607-1616. https://doi.org/10.1093/jnci/djt277.).
En resumen, el cuadro fenotípico y los criterios diagnósticos del SC siguen siendo aspectos controvertidos a pesar de los años transcurridos desde las primeras descripciones. La situación es aún más indefinida para el SBRR, síndrome del que no existen series publicadas, o para el Proteus-like, una entidad aún mal definida.
Riesgos de cáncer
El SPTH es un síndrome de predisposición al cáncer. Clásicamente estaba aceptado que los pacientes con SC tenían un riesgo elevado de padecer cáncer de mama, riesgo del 25-50%, y carcinoma folicular de tiroides, con un riesgo del 3-10%, a lo largo de la vida (Eng, 2000Eng, C. (2000). Will the real Cowden syndrome please stand up: revised diagnostic criteria. Journal of Medical Genetics, 37 (11), pp. 828-830. https://doi.org/10.1136/jmg.37.11.828.). Posteriormente se identificó el riesgo para cáncer de endometrio, que junto a los dos anteriores, componían el conjunto “clásico” de tumores asociados al SC (Eng, 2003Eng, C. (2003). PTEN: One gene, many syndromes. Human Mutation, 22 (3), pp. 183-198. https://doi.org/10.1002/humu.10257.). Durante años estos fueron los únicos riesgos reconocidos, hacia los que se dirigían todas las recomendaciones y medidas para la detección precoz.
No obstante, los estudios más recientes conceden al cáncer un papel mucho más relevante del reconocido hasta la fecha. En primer lugar, las cifras de riesgo de cáncer parecen ser superiores a las estimadas para los “cánceres clásicos”. Así, el riesgo para cáncer de mama se sitúa en el 77-85% a lo largo de la vida, el de tiroides en 35-38% y el de endometrio en el 28%. Además, existe un riesgo elevado para otros tumores como riñón (34%), colorrectal (9%) y melanoma (6%) (Nieuwenhuis et al., 2013Nieuwenhuis, M. H., Kets, C. M., Murphy-Ryan, M., Yntema, H. G., Evans, D. G., Colas, C. [...] y Vasen, H. F. A. (2013). Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Familial Cancer, 13 (1), pp. 57-63. https://doi.org/10.1007/s10689-013-9674-3.; Tan et al., 2012Tan, M. H., Mester, J. L., Ngeow, J., Ribicki, L. A., Orloff, M. S. y Eng, C. (2012). Lifetime cancer risks in individuals with germline PTEN mutations. Clinical Cancer Research, 18 (2), pp. 400-407. https://doi.org/10.1158/1078-0432.CCR-11-2283.). El riesgo a lo largo de la vida para cualquier tipo de cáncer alcanza el 85% (uno de los SPGC con mayores cifras de riesgo) y se proponen nuevas medidas de seguimiento además de la vigilancia anual de mamas, tiroides y endometrio: colonoscopia y ecografía renal, ambos con frecuencia anual a partir de los 30 años, y examen dermatológico anual desde el momento del diagnóstico (Bubien et al., 2013Bubien, V., Bonnet, F., Brouste, V., Hoppe, S., Barouk-Simonet, E., David, A. [...] y Caux, F. (2013). High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. Journal of Medical Genetics, 50, pp. 255-263. https://doi.org/10.1136/jmedgenet-2012-101339.). No obstante, algunos autores (Daniels, Rich, Weissman y Pilarsky, 2012Daniels, M. S., Rich, T., Weissman, S. y Pilarsky, R. (2012). Lifetime cancer risks of PTEN mutation carriers. Clinical Cancer Research, 18, pp. 4213-4213. https://doi.org/10.1158/1078-0432.CCR-12-0577.) han criticado la magnitud de estos riesgos, en especial de los aparecidos en el trabajo de Tan et al. (2012Tan, M. H., Mester, J. L., Ngeow, J., Ribicki, L. A., Orloff, M. S. y Eng, C. (2012). Lifetime cancer risks in individuals with germline PTEN mutations. Clinical Cancer Research, 18 (2), pp. 400-407. https://doi.org/10.1158/1078-0432.CCR-11-2283.), alegando un posible sesgo en la selección de los pacientes, por lo que podría ser prematuro recomendar cambios en la práctica clínica hasta que estas cifras no sean corroboradas por nuevos y más extensos estudios.
En resumen, los riesgos de cáncer asociados al SPTH y las medidas de detección precoz y reducción del riesgo también son aspectos no bien definidos aún, aunque parece aceptarse que existe un riesgo para una mayor variedad de tumores.
Bases moleculares
a) Gen PTEN
Localizado en 10q23.3, el gen PTEN codifica una fosfatasa que juega un papel de supresor tumoral regulando negativamente la ruta de señalización PI3K/AKT/mTOR (Maehama y Dixon, 1998Maehama, T. y Dixon, J. E. (1998). The tumor suppressor PTEN/NMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-triphosphate. Journal of Biological Chemistry, 273 (22), pp. 13375-13378. https://doi.org/10.1074/jbc.273.22.13375.). Sus funciones son parcialmente conocidas. Está implicado en el mantenimiento de la estabilidad genómica, reparación del ADN, senescencia celular y en migración y metástasis (Zhang y Yu, 2010Zhang, S. y Yu, D. (2010). PI (3) king apart PTEN´s role in cancer. Clinical Cancer Research, 16, pp. 4325-4330. https://doi.org/10.1158/1078-0432.CCR-09-2990.). Se han identificado mutaciones en PTEN en numerosas formas esporádicas de cánceres humanos (Bonneau y Longy, 2000Bonneau, D. y Longy, M. (2000). Mutations of the human PTEN gene. Human Mutation, 16 (2), pp. 109-122. https://doi.org/10.1002/1098-1004(200008)16:2<109::AID-HUMU3>3.0.CO;2-0.). Contiene nueve exones y en su estructura destacan dos dominios, el dominio fosfatasa (aa 7-185) y el C2, ambos esenciales para su función supresora tumoral, y los dominios PDZ y PEST que contribuyen a la estabilidad de la proteína (Lee et al., 1999Lee, J. O., Yang, H., Georgescu, M. M., Cristofano, A., Maehama, T., Shi, Y. [...] y Pavletich, N. P. (1999). Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell, 99 (3), pp. 323-334. https://doi.org/10.1016/S0092-8674(00)81663-3.). Liaw et al. (1997Liaw, D., Marsh, D. J., Li, J., Dahia, P. L., Wang, S. I., Zheng, Z. [...] y Parsons, R. (1997). Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nature Genetics, 16 (1), pp. 64-67. https://doi.org/10.1038/ng0597-64.) identificaron por primera vez mutaciones germinales en PTEN en varias familias con SC. Los resultados inmediatamente posteriores a esta primera descripción apuntaban a que el 80% de los casos que cumplían los criterios diagnósticos -del consorcio internacional-, presentaban mutación germinal en PTEN. Este porcentaje ha ido disminuyendo en la medida en que se ha analizado un mayor número de casos, situándose en la actualidad entre el 30 y el 35% de los pacientes que satisfacen los criterios clínicos (Pilarski et al., 2013Pilarski, R., Burt, R., Kohlman, W., Pho, L., Shannon, K. M. y Swisher, E. (2013). Cowden Syndrome and the PTEN Hamartoma Tumor Syndrome: Systematic review and revised diagnostic criteria. Journal of the National Cancer Institute, 105 (21), pp. 1607-1616. https://doi.org/10.1093/jnci/djt277.; Tan et al., 2011Tan, M. H., Mester, J., Peterson, C., Yang, Y., Chen, J. L., Rybicki, L. A [...] y Eng, C. (2011). A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. The American Journal of Human Genetics, 88 (1), pp. 42-56. https://doi.org/10.1016/j.ajhg.2010.11.013.). Las mutaciones se distribuyen a lo largo de todo el gen, incluyendo la región promotora, con un claro punto caliente entre los exones cinco y ocho, y siendo raras las mutaciones entre los exones uno al cuatro y en el nueve (Hollander, Blumenthal y Dennis, 2011Hollander, M. C., Blumenthal, G. M. y Dennis, P. A. (2011). PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nature Reviews Cancer, 11 (4), pp. 289-301. https://doi.org/10.1038/nrc3037.). Grandes reordenamientos del gen se han descrito; algunos de ellos son extensas microdeleciones en la región 10q22-q23 que implican pérdidas de PTEN y de BMPR1A y pueden asociarse a fenotipos específicos (Delnatte et al., 2006Delnatte, C., Sanlaville, D., Mougenot, J. F., Vermeexch, J. R., Houdayer, C., Blois, M. C. de [...] y Stoppa-Lyonnet, D. (2006). Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. The American Journal of Human Genetics, 78 (6), pp. 1066-1074. https://doi.org/10.1086/504301.). Tomando como base solo los resultados de tres series de pacientes, que sumaron más de 100 casos con mutación germinal en PTEN (dos estudios europeos y uno de EEUU), no se ha apreciado una clara correlación genotipo-fenotipo (Bubien et al., 2013Bubien, V., Bonnet, F., Brouste, V., Hoppe, S., Barouk-Simonet, E., David, A. [...] y Caux, F. (2013). High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. Journal of Medical Genetics, 50, pp. 255-263. https://doi.org/10.1136/jmedgenet-2012-101339.; Nieuwenhuis et al., 2013Nieuwenhuis, M. H., Kets, C. M., Murphy-Ryan, M., Yntema, H. G., Evans, D. G., Colas, C. [...] y Vasen, H. F. A. (2013). Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Familial Cancer, 13 (1), pp. 57-63. https://doi.org/10.1007/s10689-013-9674-3.; Tan et al., 2012Tan, M. H., Mester, J. L., Ngeow, J., Ribicki, L. A., Orloff, M. S. y Eng, C. (2012). Lifetime cancer risks in individuals with germline PTEN mutations. Clinical Cancer Research, 18 (2), pp. 400-407. https://doi.org/10.1158/1078-0432.CCR-11-2283.).
b) Otros genes
En un alto porcentaje (65-70%) de los pacientes con sospecha de SPTH no se detecta mutación germinal en PTEN. Teniendo en cuenta que tanto del SC como el SBRR son entidades que se definen y diagnostican por sus signos clínicos, mientras que la presencia de una mutación germinal en PTEN es condición indispensable para diagnosticar de SPTH, la no detección de la mutación en un paciente diagnosticado de SC o de SBRR crea notables incertidumbres. ¿La ausencia de mutación invalida el diagnóstico de SC o de SBRR?, ¿son recomendables, y con qué pautas, las medidas de seguimiento y prevención en un paciente diagnosticado de SC sin mutación detectable en PTEN? Mientras tratamos de encontrar respuestas razonables a estas preguntas, cabe mencionar que se han descrito otros mecanismos genéticos y otros genes como posibles responsables del síndrome. Alimonti et al. (2010Alimonti, A., Carracedo, A., Clohessy, J. G., Trotman, L. C., Nardella, C., Egia, A. [...] y Richardson, A. L. (2010). Subtle variations in Pten dose determine cancer susceptibility. Nature Genetics, 42 (5), pp. 454-458. https://doi.org/10.1038/ng.556.) demostraron que ciertos cambios sutiles en los niveles de expresión de PTEN pueden aumentar sustancialmente el riesgo de desarrollar cáncer de mama. Ratones que manifestaban niveles de PTEN del 80% tenían un riesgo del 40% de tener cáncer de mama. No hay resultados en humanos que avalen los obtenidos en ratones por estos autores.
La hipermetilación germinal del promotor del gen KLLN se ha propuesto recientemente como causa del SPTH. KLLN comparte promotor con PTEN y la hipermetilación del mismo reduce 250 veces la expresión de KLLN y contrariamente a lo esperable, incrementa ligeramente la expresión de PTEN, en el 37% de los pacientes con SC y estudio negativo del gen PTEN (Cho y Liang, 2008Cho, Y. J. y Liang, P. (2008). KILLIN is a p53-regulated nuclear inhibitor of DNA synthesis. Proceedings of the National Academy of Sciences, 105 (14), pp. 5396-5401. https://doi.org/10.1073/pnas.0705410105.). El uso de agentes demetilantes en líneas linfoblásticas de estos pacientes restaura la expresión de KLLN (Bennett et al., 2011Bennett, K. L., Campbell, R., Ganapathi, S., Zhou, M., Rini, B., Ganapathi, R. [...] y Eng, C. (2011). Germline and somatic DNA methylation and epigenetic regulation of KILLIN in renal cell carcinoma. Genes, Chromosomes and Cancer, 50 (8), pp. 654-661. https://doi.org/10.1002/gcc.20887.). No se dispone de confirmación de estos resultados en otras series de pacientes distintas a las del grupo de la dra. C. Eng de Cleveland, Ohio (Cho y Liang, 2008Cho, Y. J. y Liang, P. (2008). KILLIN is a p53-regulated nuclear inhibitor of DNA synthesis. Proceedings of the National Academy of Sciences, 105 (14), pp. 5396-5401. https://doi.org/10.1073/pnas.0705410105.). También este mismo grupo ha descrito variantes en los genes SDHB, SDHD del complejo II de la cadena respiratoria mitocondrial, potencialmente implicados en el 10% de los pacientes con SC o SC-like con estudio negativo de PTEN/KLLN (Ni y Eng, 2012Ni, Y. y Eng, C. (2012). Vitamin E protects against lipid peroxidation and rescues tumorigenic phenotypes in cowden/cowden-like patient-derived lymphoblast cells with germline SDHx variants. Clinical Cancer Research, 18, pp. 4954-4961. https://doi.org/10.1158/1078-0432.CCR-12-1055.), si bien algunas de las variantes encontradas son frecuentes en población general por lo que estos resultados requieren confirmación en nuevas series. Y por último, ese grupo de investigadores también ha sido el primero, y de momento el único, que ha descrito mutaciones germinales en AKT1 y PI3KCA, ambos genes miembros de la misma ruta de señalización que PTEN, en una pequeña proporción de pacientes con SC o SC-like (Orloff et al., 2013Orloff, M. S., He, X., Peterson, C., Chen, F., Chen, J. L., Mester, J. L. y Eng, C. (2013). Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. The American Journal of Human Genetics, 92 (1), pp. 76-80. https://doi.org/10.1016/j.ajhg.2012.10.021.).
Manejo de pacientes y expectativas terapéuticas
El SPTH es un síndrome difícil de identificar, como ya se ha mencionado, que con frecuencia se diagnostica tarde y, en más de un tercio de los casos, después de haber estudiado otros genes de susceptibilidad, principalmente BRCA1/2 y APC. Existen grandes sesgos a la hora de remitir pacientes para el estudio del gen PTEN en función de la especialidad clínica del profesional sanitario que sospecha el síndrome. El conocimiento está muy parcelado. Aunque es un síndrome raro (alrededor de un caso cada 200.000 nacimientos) algunos autores opinan que puede existir un infradiagnóstico por la gran variación fenotípica que presenta (Eng, 2000Eng, C. (2000). Will the real Cowden syndrome please stand up: revised diagnostic criteria. Journal of Medical Genetics, 37 (11), pp. 828-830. https://doi.org/10.1136/jmg.37.11.828.). En cualquier caso, ante la sospecha del mismo, los pacientes deberían ser remitidos a consultas de consejo genético especializadas, en las que llevar a cabo la prueba genética, coordinar el seguimiento del paciente e iniciar el estudio familiar. Las recomendaciones de seguimiento, muy centradas en el cáncer, han sido revisadas recientemente en el trabajo de Bubien et al. (2013Bubien, V., Bonnet, F., Brouste, V., Hoppe, S., Barouk-Simonet, E., David, A. [...] y Caux, F. (2013). High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. Journal of Medical Genetics, 50, pp. 255-263. https://doi.org/10.1136/jmedgenet-2012-101339.), y aparecen en la tabla 4.
|
Tabla 4. Recomendaciones para el seguimiento del síndrome de PTEN-tumores hamartomasos
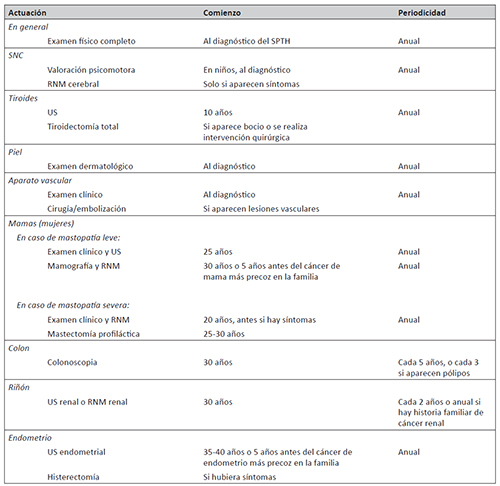
* RNM: resonancia nuclear magnética. SNC: Sistema nervioso central US: Ultrasonidos.
Fuente: Elaboración propia a partir de Bubien et al., 2013.
[Descargar tamaño completo]
| |
Respecto al tratamiento del SC, el desarrollo de numerosos inhibidores de varios nodos en la ruta PI3K/AKT/mTOR (inhibidores de AKT, inhibidores Pan-PI3K, inhibidores mTORC1, etc., revisado por Dienstmann, Rodon, Serra y Tabernero, 2014Dienstmann, R., Rodon, J., Serra, V. y Tabernero, J. (2014). Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Molecular Cancer Therapeutics, 13 (5), pp. 1021-1031. https://doi.org/10.1158/1535-7163.MCT-13-0639.), ha abierto notables expectativas. En la reunión de ASCO 2013, Komiya y sus colegas presentaron los resultados de un estudio piloto en el que 18 pacientes con SC fueron tratados con 2mg diarios de sirolimus, durante dos meses. Los pacientes experimentaron una mejoría de sus síntomas gastrointestinales, mucocutáneos y de su función cerebelar (Komiya et al., 2013Komiya, T., Blumenthal, G. M., Ballas, M. S., Dechowdhury, R., Manu, M., Fioravanti, S. [...] y Hewitt, S. M. (2013). A pilot study of sirolimus (S) in subjects with Cowden syndrome (CS) with germ-line mutations in PTEN. Journal of Clinical Oncology, 31 (15_suppl), pp. 2532-2532.). Sin embargo, a fecha de hoy estos resultados no se han publicado, ni tampoco resultados de otras series con análogos del sirolimus como el everolimus o el temsirolimus. En consecuencia, no se dispone aún de datos contrastados que avalen las buenas expectativas iniciales para el tratamiento de los pacientes con inhibidores PI3K/AKT/mTOR.
En definitiva y a modo de resumen, el SPTH es una entidad difícil de reconocer, con un diagnóstico que suele retrasarse, con un fenotipo muy variable, aún no totalmente definido. Los criterios clínicos recientemente revisados han identificado una serie de características posiblemente sobrevaloradas, frente a otras con mayor valor predictivo de cara a la prueba genética. El uso de estos nuevos criterios clínicos podría ayudar a reconocer de manera más precisa los candidatos al estudio genético, y a acortar el tiempo hasta el diagnóstico. Los pacientes portadores de mutaciones en PTEN parecen tener un alto riesgo de padecer cáncer (en torno al 85% de manera global), incluyendo una amplia variedad de tumores (mama, tiroides, endometrio, riñón, colorrectal y melanoma). Estas nuevas estimaciones del riesgo de cáncer conllevan modificaciones en las recomendaciones de seguimiento. Desde el punto de vista genético, el síndrome es heterogéneo y se han descrito hasta seis genes distintos potencialmente implicados, cuyo papel requiere confirmación en series más amplias de pacientes y en distintas poblaciones. Pero además, en un porcentaje muy importante de pacientes no se identifica la mutación causal por lo que es muy probable la existencia de otros genes de susceptibilidad, cuya identificación es necesaria. Los inhibidores de la ruta PI3K/Akt/mTOR han abierto nuevas y prometedoras vías en el tratamiento de los pacientes, aunque por el momento no hay datos contrastados disponibles.
HERRAMIENTAS PARA LA BÚSQUEDA DE NUEVOS GENES IMPLICADOS EN LOS SPGC Top
Como se ha mencionado anteriormente el número de síndromes de susceptibilidad al cáncer es elevado, mas de 200, y uno de los principales problemas que tiene su abordaje es, por un lado, la gran heterogeneidad genética que presentan la mayoría de ellos y, por otro, el desconocimiento de las bases genéticas en muchos de estos síndromes. Esto hace que gran parte de los estudios sean incompletos y por consiguiente el consejo genético sea de poco valor para la familia.
Los cánceres más frecuentes, como el de mama, colon, próstata, pulmón, páncreas y una larga lista, son buenos ejemplos de lo anterior. El cáncer de mama solo tiene dos genes conocidos de alta penetración, BRCA1 y BRCA2, que explican alrededor de un 20% de las familias con cáncer de mama y ovario, y un tercer gen recientemente descubierto, RAD51C, que explicaría un porcentaje del 1% de las familias (Meindl et al., 2010Meindl, A., Hellebrand, H., Wiek, C., Erven, V., Wappenschmidt, B., Niederacher, D. [...] y Hanenberg, H. (2010). Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nature Genetics, 42 (5), pp. 410-414. https://doi.org/10.1038/ng.569.; Osorio et al., 2012Osorio, A., Endt, D., Fernández, F., Eirich, K., Hoya, M. de la, Schmutzler, R. [...] y Benítez, J. (2012). Predominance of pathogenic missense variants in the RAD51C gene occurring in breast and ovarian cancer families. Human Molecular Genetics, 21 (13), pp. 2889-2898. https://doi.org/10.1093/hmg/dds115.). Hay además otro conjunto de genes de moderada penetración, entre los que se encuentran PALB2, BRIP1, RAD51D, CHEK2 y ATM (Filippini y Vega, 2013Filippini, S. E. y Vega, A. (2013). Breast cancer genes: beyond BRCA1 and BRCA2. Frontiers in Bioscience, 18, pp. 1358-1372. https://doi.org/10.2741/4185.), que explicarían menos del 5% de las familias. De esta manera y con el conocimiento actual que tenemos, solo el 25-30% de las familias con cáncer de mama y ovario tendrían una base genética conocida. Más complicado es el caso del cáncer de próstata o pulmón donde no hay genes descritos que puedan explicar un mínimo porcentaje de familias con estos cánceres.
Por otro lado, nos encontramos con síndromes de susceptibilidad muy poco frecuentes. Estamos hablando de incidencias menores del 1/100.000 que hacen muy difícil identificar el gen asociado a la enfermedad, salvo que se den circunstancias favorables. Algunos síndromes de inmunodeficiencia severa o de malformaciones asociadas a cáncer son buenos ejemplos.
Finalmente, una tercera situación la constituyen los cánceres que se explicarían tomando como base un modelo poligénico, o mejor multifactorial, entre los que el cáncer de vejiga o de esófago representan buenos modelos. En este último existe una condición previa que es el esófago de Barrett (EB), una patología premaligna que se caracteriza porque el epitelio escamoso del esófago es reemplazado por el epitelio columnar dando lugar a una metaplasia que puede llegar a degenerar en circunstancias habituales (reflujo esofágico, hernia de hiato) en un adenocarcinoma de esófago (Pascarenco et al., 2014Pascarenco, O. D., Boeriu, A., Mocan, S., Pascarenco, G., Drasoveanu, S., Galeanu, M. y Dobru, D. (2014). Barrett’s esophagus and intestinal metaplasia of gastric cardia: prevalence, clinical, endoscopic and histological features. Journal of Gastrointestinal and Liver Diseases, 23 (1), pp. 19-25.). El EB está presente en muchas familias en forma de agregación, o con otros miembros con hernia o con reflujo o incluso con cáncer de esófago, pero sin que hasta el momento se haya identificado un gen de susceptibilidad.
Genes de susceptibilidad al cáncer
Hasta hace poco se pensaba que los casos esporádicos se podían explicar tomando como base los modelos poligénicos donde un conjunto de genes de baja susceptibilidad, con riesgos (OR) <1,5, se combinarían según un modelo multiplicativo, dando lugar a un incremento importante de riesgo final. Las nuevas técnicas de genotipación masiva han demostrado que esto es correcto y, de hecho, se han realizado desde 2010 una gran cantidad de estudios de genotipación masiva, los llamados GWAS (Genome Wide Association Studies), en los que comparando miles de casos y miles de controles se ha podido confirmar la existencia de estos genes de una manera robusta y sólida (Milne y Benítez, 2008Milne, R. L. y Benítez, J. (2008). Current strategies in the search for low penetrance genes in cancer. Histology and Histopathology, 23 (4), pp. 507-514.). Así, se han identificado mediante estudios de asociación 67 genes de baja penetración en cáncer de mama, 12 genes en cáncer de colon, 5 genes en cáncer de pulmón o 27 genes en cáncer de próstata. Todos ellos tienen unos OR<1,5 y además contribuyen a explicar un porcentaje de familias con esos diferentes tipos de cáncer. En mama hasta un 27% del riesgo de cáncer familiar, un 7% en colon y pulmón y hasta un 30% en próstata (Ghoussaini, Pharoah e Easton, 2013Ghoussaini, M., Pharoah, P. D. y Easton, D. F. (2013). Inherited genetic susceptibility to breast cancer: the beginning of the end or the end of the beginning?. The American Journal of Pathology, 183 (4), pp. 1038-1051. https://doi.org/10.1016/j.ajpath.2013.07.003.) (figura 1). Estos resultados vienen a apoyar la hipótesis de que parte de las familias con cáncer pueden explicarse tomando como base un modelo poligénico, posiblemente con un gen mayor confiriendo un OR superior a dos o tres, y otro conjunto de genes de baja penetración adicionales completando el espectro de alto riesgo. Este hecho explicaría en parte la dificultad que existe para encontrar nuevos genes de alta susceptibilidad en mama (BRCAX) o en colon (HNPCCX).
|
Figura 1. Esquema donde se muestra la localización de muchas de las variantes de baja susceptibilidad identificadas en diferentes cánceres esporádicos mediante estudios de GWAS. Estas variantes contribuyen también al riesgo de cáncer familiar

Fuente: Ghoussaini. Pharoah e Easton, 2013.
[Descargar tamaño completo]
| |
La secuenciación masiva del exoma
Desde hace años hemos asistido a una nueva revolución tecnológica. La posibilidad de secuenciar todos nuestros genes de una forma rápida (pocos días) y económica (menos de 1.000 euros) es lo que conocemos como secuenciación masiva del exoma (WES) y, yendo más allá, tenemos también la posibilidad de secuenciación masiva del genoma. La diferencia entre uno y otro es la información que se obtiene, ya que en el primer caso se secuencian exclusivamente los exones de un gen, la zona codificante, mientras que en el segundo caso se incluyen además las regiones intrónicas y las regiones intergénicas, es decir, todo nuestro genoma versus el 2% del mismo que es donde se acumulan el 85-90% de las mutaciones. Debido a la complejidad de esta tecnología especialmente en lo que al análisis y al almacenamiento de datos generados se refiere, la WES es el método utilizado en la actualidad para la búsqueda de nuevos genes, que es su aplicación más importante.
La WES ha venido a cubrir una importante parcela en la búsqueda de genes responsables de enfermedades genéticas en general y en el cáncer familiar en particular. La estrategia es la misma, secuenciar toda la zona codificante con las diferentes plataformas comerciales que hay en la actualidad (Illumina, Applied, Roche), y para ello se parte de una fragmentación del genoma en pequeños fragmentos de 150-400 pares de base; a continuación hay que capturar los fragmentos pertenecientes a los exones de los genes, o simplemente los genes incluyendo exones e intrones, secuenciar esos miles de fragmentos, y posteriormente ensamblarlos en un continuo donde se pueda leer de forma ordenada el genoma o exoma y analizar si existen mutaciones puntuales, deleciones, inserciones u otras alteraciones que puedan explicar la susceptibilidad al cáncer en esas familias. Este proceso es complejo, aunque actualmente está informatizado y automatizado en los diferentes pipelines bioinformáticos que se utilizan en los distintos laboratorios. Estas herramientas permiten alinear las secuencias que se generan, identificar los cambios en las secuencias comparándolos con las secuencias de referencia, pero después hay que eliminar los falsos positivos, hay que cruzar los datos con aquellos que se encuentran en las diferentes bases de datos, y mediante estos y otros filtros hay que dejar en el camino 150.000 variantes por término medio que son las que se obtienen inicialmente y quedarse con la media docena de variantes o genes que pueden ser los realmente de interés que estamos buscando (Tucker, Marra y Friedman, 2009Tucker, T., Marra, M. y Friedman, J. M. (2009). Massively parallel sequencing: the next big thing in genetic medicine. The American Journal of Human Genetics, 85 (2), pp. 142-154. https://doi.org/10.1016/j.ajhg.2009.06.022.) (Véase figura 2).
|
Figura 2. Esquema simplificado del proceso de secuenciación masiva. A la izquierda las fases de secuenciación y a la derecha el pipeline del análisis de datos con algunos de los softwares que se utilizan para el análisis
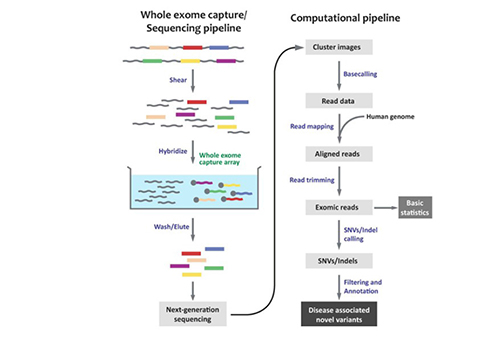
Fuente: Elaboración propia.
[Descargar tamaño completo]
| |
Resultados de la secuenciación masiva en la búsqueda de nuevos genes
La secuenciación masiva ha puesto de manifiesto dos aspectos. El primero es que la identificación de nuevos genes no es una tarea tan sencilla como lo que parecía originalmente. La segunda es que nuestro genoma o exoma es mucho más complejo de lo que suponíamos y que en un gran número de familias el cáncer se debe a la interacción de muchos genes que darán finalmente un riesgo similar al de un gen de alta susceptibilidad.
En el primer caso los genes que se han ido identificando corresponden a síndromes asociados, en su mayoría a modelos recesivos o poco frecuentes, o en otras ocasiones formando parte de ese espectro de genes de heterogeneidad genética. MAX fue el primer gen descrito por secuenciación masiva en cáncer familiar en el año 2011 y nuestra Unidad del CIBERER (U706) tuvo ese honor (Comino-Méndez et al., 2011Comino-Méndez, I, Gracia-Aznárez, F. J., Schiavi, F., Landa, I., Leandro-García, L. J., Letón, R. [...] y Cascón, A. (2011). Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nature Genetics, 43 (7), pp. 663-667. https://doi.org/10.1038/ng.861.). Este es un gen que explica menos de un 5% de familias con feocromocitoma y paragangiomas y también un 1% de las formas esporádicas. En el 2012 tres nuevos genes, uno en cáncer de páncreas que explicaba un 2% de familias (ATM) (Roberts et al., 2012Roberts, N. J., Jiao, Y., Yu, J., Kopolovich, L., Petersen, G. M., Bondy, M. L. [...] y Klein, A. P. (2012). ATM mutations in patients with hereditary pancreatic cancer. Cancer Discovery, 2, pp. 41-46. https://doi.org/10.1158/2159-8290.CD-11-0194.), otro en una forma rara de paragangliomas con policitemia (HIF2A) (Zhuang et al., 2012Zhuang, Z., Yang, C., Lorenzo, F., Merino, M., Fojo, T., Kebebew, E. [...] y Pacak, K. (2012). Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. The New England Journal of Medicine, 367 (10), pp. 922-930. https://doi.org/10.1056/NEJMoa1205119.) y el tercero en esófago (RHBDF2) (Blaydon et al., 2012Blaydon, D. C., Etheridge, S. L., Risk, J. M., Hennies, H. C., Gay, L. J., Carroll, R. [...] y Bishop, D. T. (2012). RHBDF2 mutations are associated with tylosis, a familial esophageal cancer syndrome. The American Journal of Human Genetics, 90 (2), pp. 340-346. https://doi.org/10.1016/j.ajhg.2011.12.008.), fueron descritos. En el 2013 un nuevo gen de disqueratosis congénita (RTL1) (Ballew et al., 2013Ballew, B. J., Yeager, M., Jacobs, K., Giri, N., Boland, J., Burdett, L. [...] y Savage, S. A. (2013). Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in dyskeratosis congenita. Human Genetics, 132 (4), pp. 473-480. https://doi.org/10.1007/s00439-013-1265-8.) y otro de Anemia de Fanconi (FANCQ) (Bogliolo et al., 2013Bogliolo, M., Schuster, B., Stoepker, C., Derkunt, B., Su, Y., Raams, A. [...] y Casado, J. A. (2013). Mutations in ERCC4, Encoding the DNA-Repair Endonuclease XPF, Cause Fanconi Anemia. The American Journal of Human Genetics, 92 (5), pp. 800-806. https://doi.org/10.1016/j.ajhg.2013.04.002.), fueron publicados, y finalmente en el 2014 varios trabajos en distintos tipos de cáncer familiar, melanoma y POT1, cáncer de pulmón y HER2, y en cáncer de ovario de célula pequeña (Robles-Espinoza et al., 2014Robles-Espinoza, C. D., Harland, M., Ramsay, A. J., Aoude, L. G., Quesada, V., Ding, Z. [...] y Adams, D. J. (2014). POT1 loss-of-function variants predispose to familial melanoma. Nature Genetics, 46 (5), pp. 478-481. https://doi.org/10.1038/ng.2947.; Shi et al., 2014Shi, J., Yang, X. R., Ballew, B., Rotunno, M., Calista, D., Fargnoli, M. C. [...] y Landi, M. T. (2014). Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nature Genetics, 46 (5), pp. 482-486. https://doi.org/10.1038/ng.2941.), han sido los identificados hasta el momento. Como se puede apreciar el número de genes es escaso, dada la dificultad de encontrar familias extensas y la necesidad de hacer gran cantidad de estudios funcionales para demostrar que se trata del gen responsable.
Más complicado y complejo es el abordaje de otros cánceres familiares más comunes como son el de mama o el de colon. Los estudios realizados en los primeros con diferentes aproximaciones, a saber: a) en familias con un mínimo de ocho a diez mujeres con cáncer de mama en diversas generaciones (Gracia-Aznarez et al., 2013Gracia-Aznarez, F. J., Fernández, V., Pita, G., Peterlongo, P., Domínguez, O., Hoya, M. de la [...] y Benítez, J. (2013). Whole exome sequencing suggests much of non-BRCA1/BRCA2 familial breast cancer is due to moderate and low penetrance susceptibility alleles. PLoS One, 8 (2), e55681. https://doi.org/10.1371/journal.pone.0055681.), b) en regiones del genoma previamente identificadas como candidatas mediante estudios de ligamientos (Hilbers et al., 2013Hilbers, F. S., Meijers, C. M., Laros, J. F., Galen, M. van, Hoogerbrugge, N., Vase, H. F. A. [...] y Devilee, P. (2013). Exome sequencing of germline DNA from non-BRCA1/2 familial breast cancer cases selected on the basis of aCGH tumor profiling. PLoS One, 8 (1), e55734. https://doi.org/10.1371/journal.pone.0055734.; Rosa-Rosa et al., 2010Rosa-Rosa, J. M., Gracia-Aznárez, F. J., Hodges, E., Pita, G., Rooks, M., Xuan, Z. [...] y Benítez, J. (2010). Deep sequencing of target linkage assay-identified regions in familial breast cancer: methods, analysis pipeline and troubleshooting. PLoS One, 5 (4), e9976. https://doi.org/10.1371/journal.pone.0009976.), c) en múltiples familias con tres o cuatro casos de cáncer de mama (Snape et al., 2012Snape, K., Ruark, E., Tarpey, P., Renwick, A., Turnbull, C., Seal, S. [...] y Rahman, N. (2012). Predisposition gene identification in common cancers by exome sequencing: insights from familial breast cancer. Breast Cancer Research Treatement, 134 (1), pp. 429-433. https://doi.org/10.1007/s10549-012-2057-x.; Thompson et al., 2012Thompson, E. R., Doyle, M. A., Ryland, G. L., Rowley, S. M., Choong, D. Y., Tothill, R. W. [...] y Campbell, I. G. (2012). Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genetics, 8 (9), e1002894. https://doi.org/10.1371/journal.pgen.1002894.), han mostrado en todas las ocasiones la ausencia de un gen de alta susceptibilidad que explicara un bajo porcentaje de familias o incluso una sola de las grandes familias poniendo de manifiesto la importancia que tienen los genes de moderada y baja susceptibilidad para explicar su origen (Lu et al., 2014Lu, Y., Ek, W. E., Whiteman, D., Vaughan, T. L., Spurdle, A. B., Easton, D. F. [...] y Macgregor, S. (2014). Most common ‘sporadic’ cancers have a significant germline genetic component. Human Molecular Genetics, 23 (22), pp. 6112-6118. https://doi.org/10.1093/hmg/ddu312.). Lo mismo ocurre en el cáncer de colon familiar, ya que los intentos que ha habido de buscar nuevos genes HNPCCX en grandes familias han identificado únicamente posibles variantes que puedan jugar algún papel.
La secuenciación masiva en estas familias ha mostrado una nueva categoría de variantes, las variantes raras, aquellas variantes que aparecen al secuenciar en exones y que generan un cambio de aminoácido o afectan al splicing, que no han sido descritas nunca ni se encuentran en bases de datos, y que pueden ayudar a explicar esta mayor susceptibilidad. La única forma de entender el significado de estas variantes raras es mediante estudios poblacionales, bien de amplios estudios de asociación entre casos y controles o bien mediante secuenciación del gen donde se encuentra la variante en un amplio número de casos y de controles. En este sentido se puso recientemente en marcha un consorcio en cáncer de mama cuyo nombre es COMPLEXO (Southey et al., 2013Southey, M. C., Park, D. J., Nguyen-Dumont, T., Campbell, I., Thompson, E., Trainer, A. H. [...] y Goldgar, D. E. (2013). COMPLEXO: identifying the missing heritability of breast cancer via next generation collaboration. Breast Cancer Ressearch, 15 (3), pp. 402-402. https://doi.org/10.1186/bcr3434.), formado por una veintena de grupos de todo el mundo con el objetivo de dar respuesta a esta situación.
CONCLUSIONES Top
El cáncer hereditario, junto con los síndromes de susceptibilidad al cáncer, constituye una entidad compleja ya que la mayoría de los esfuerzos se han centrado en los cánceres más comunes (media docena) dejando de lado las restantes 200 formas. Hay una importante tarea clínica, diagnóstica, preventiva y terapéutica por hacer, que la genética empieza a analizar en la actualidad, en algunos casos a entender, y en otros a descubrir. La mayor formación en esta área, que se está recibiendo fuera de los currículos oficiales, está contribuyendo a ello. El incremento en el número de los grupos de investigación ha sido el gran detonante, y finalmente las nuevas tecnologías nos enseñan, a la par que facilitan la explicación de determinados interrogantes que se llevan tratando de descifrar desde hace tiempo. Los próximos años van a ser productivos y se va a poder trasladar gran parte de los desarrollos diagnósticos y terapéuticos al estudio del paciente.
BIBLIOGRAFÍATop
|
| ○ | Alimonti, A., Carracedo, A., Clohessy, J. G., Trotman, L. C., Nardella, C., Egia, A. [...] y Richardson, A. L. (2010). Subtle variations in Pten dose determine cancer susceptibility. Nature Genetics, 42 (5), pp. 454-458. https://doi.org/10.1038/ng.556 |
| ○ | American Cancer Society (2018). Cancer Facts & Figures 2018. Atlanta, Ga: American Cancer Society. |
| ○ | Ballew, B. J., Yeager, M., Jacobs, K., Giri, N., Boland, J., Burdett, L. [...] y Savage, S. A. (2013). Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in dyskeratosis congenita. Human Genetics, 132 (4), pp. 473-480. https://doi.org/10.1007/s00439-013-1265-8 |
| ○ | Bennett, K. L., Campbell, R., Ganapathi, S., Zhou, M., Rini, B., Ganapathi, R. [...] y Eng, C. (2011). Germline and somatic DNA methylation and epigenetic regulation of KILLIN in renal cell carcinoma. Genes, Chromosomes and Cancer, 50 (8), pp. 654-661. https://doi.org/10.1002/gcc.20887 |
| ○ | Bennett, R. L., Steinhaus, K. A., Uhrich, S. B., O’Sullivan, C. K., Resta, R. G., Lochner-Doyle, D. [...] y Hamanishi, J. (1995). Recommendations for standardized human pedigree nomenclature. The American Journal of Human Genetics, 56, pp. 745-752. |
| ○ | Blaydon, D. C., Etheridge, S. L., Risk, J. M., Hennies, H. C., Gay, L. J., Carroll, R. [...] y Bishop, D. T. (2012). RHBDF2 mutations are associated with tylosis, a familial esophageal cancer syndrome. The American Journal of Human Genetics, 90 (2), pp. 340-346. https://doi.org/10.1016/j.ajhg.2011.12.008 |
| ○ | Bogliolo, M., Schuster, B., Stoepker, C., Derkunt, B., Su, Y., Raams, A. [...] y Casado, J. A. (2013). Mutations in ERCC4, Encoding the DNA-Repair Endonuclease XPF, Cause Fanconi Anemia. The American Journal of Human Genetics, 92 (5), pp. 800-806. https://doi.org/10.1016/j.ajhg.2013.04.002 |
| ○ | Bonneau, D. y Longy, M. (2000). Mutations of the human PTEN gene. Human Mutation, 16 (2), pp. 109-122. https://doi.org/10.1002/1098-1004(200008)16:2<109::AID-HUMU3>3.0.CO;2-0 |
| ○ | Bubien, V., Bonnet, F., Brouste, V., Hoppe, S., Barouk-Simonet, E., David, A. [...] y Caux, F. (2013). High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. Journal of Medical Genetics, 50, pp. 255-263. https://doi.org/10.1136/jmedgenet-2012-101339 |
| ○ | Cho, Y. J. y Liang, P. (2008). KILLIN is a p53-regulated nuclear inhibitor of DNA synthesis. Proceedings of the National Academy of Sciences, 105 (14), pp. 5396-5401. https://doi.org/10.1073/pnas.0705410105 |
| ○ | Comino-Méndez, I, Gracia-Aznárez, F. J., Schiavi, F., Landa, I., Leandro-García, L. J., Letón, R. [...] y Cascón, A. (2011). Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nature Genetics, 43 (7), pp. 663-667. https://doi.org/10.1038/ng.861 |
| ○ | Daniels, M. S., Rich, T., Weissman, S. y Pilarsky, R. (2012). Lifetime cancer risks of PTEN mutation carriers. Clinical Cancer Research, 18, pp. 4213-4213. https://doi.org/10.1158/1078-0432.CCR-12-0577 |
| ○ | Delnatte, C., Sanlaville, D., Mougenot, J. F., Vermeexch, J. R., Houdayer, C., Blois, M. C. de [...] y Stoppa-Lyonnet, D. (2006). Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. The American Journal of Human Genetics, 78 (6), pp. 1066-1074. https://doi.org/10.1086/504301 |
| ○ | Dienstmann, R., Rodon, J., Serra, V. y Tabernero, J. (2014). Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Molecular Cancer Therapeutics, 13 (5), pp. 1021-1031. https://doi.org/10.1158/1535-7163.MCT-13-0639 |
| ○ | Eng, C. (2001). PTEN Hamartoma Tumor Syndrome (PHTS). GeneReviews at GeneTests: Medical Genetics Information Resource. [En línea]. Disponible en https://www.ncbi.nlm.nih.gov/books/NBK1488/ |
| ○ | Eng, C. (2000). Will the real Cowden syndrome please stand up: revised diagnostic criteria. Journal of Medical Genetics, 37 (11), pp. 828-830. https://doi.org/10.1136/jmg.37.11.828 |
| ○ | Eng, C. (2003). PTEN: One gene, many syndromes. Human Mutation, 22 (3), pp. 183-198. https://doi.org/10.1002/humu.10257 |
| ○ | Faller, H., Schuler, M., Richard, M., Heckl, U., Weis, J. y Küffner, R. (2013). Effects of psycho-oncologic interventions on emotional distress and quality of life in adult patients with cancer: systematic review and meta-analysis. Journal of Clinical Oncology, 31 (6), pp. 782-793. https://doi.org/10.1200/JCO.2011.40.8922 |
| ○ | Filippini, S. E. y Vega, A. (2013). Breast cancer genes: beyond BRCA1 and BRCA2. Frontiers in Bioscience, 18, pp. 1358-1372. https://doi.org/10.2741/4185 |
| ○ | Frank, S. A. (2004). Genetic predisposition to cancer - insights from population genetics. Nature Reviews Genetics, 5 (10), pp. 764-772. https://doi.org/10.1038/nrg1450 |
| ○ | Ghoussaini, M., Pharoah, P. D. y Easton, D. F. (2013). Inherited genetic susceptibility to breast cancer: the beginning of the end or the end of the beginning?. The American Journal of Pathology, 183 (4), pp. 1038-1051. https://doi.org/10.1016/j.ajpath.2013.07.003 |
| ○ | Gorlin, R. J., Cohen Jr., M. M., Condon, L. M. y Burke, B. A. (1992). Bannayan-Riley-Ruvalcaba syndrome. American Journal of Medical Genetics, 44 (3), pp. 307-314. https://doi.org/10.1002/ajmg.1320440309 |
| ○ | Gracia-Aznarez, F. J., Fernández, V., Pita, G., Peterlongo, P., Domínguez, O., Hoya, M. de la [...] y Benítez, J. (2013). Whole exome sequencing suggests much of non-BRCA1/BRCA2 familial breast cancer is due to moderate and low penetrance susceptibility alleles. PLoS One, 8 (2), e55681. https://doi.org/10.1371/journal.pone.0055681 |
| ○ | Hilbers, F. S., Meijers, C. M., Laros, J. F., Galen, M. van, Hoogerbrugge, N., Vase, H. F. A. [...] y Devilee, P. (2013). Exome sequencing of germline DNA from non-BRCA1/2 familial breast cancer cases selected on the basis of aCGH tumor profiling. PLoS One, 8 (1), e55734. https://doi.org/10.1371/journal.pone.0055734 |
| ○ | Hollander, M. C., Blumenthal, G. M. y Dennis, P. A. (2011). PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nature Reviews Cancer, 11 (4), pp. 289-301. https://doi.org/10.1038/nrc3037 |
| ○ | Komiya, T., Blumenthal, G. M., Ballas, M. S., Dechowdhury, R., Manu, M., Fioravanti, S. [...] y Hewitt, S. M. (2013). A pilot study of sirolimus (S) in subjects with Cowden syndrome (CS) with germ-line mutations in PTEN. Journal of Clinical Oncology, 31 (15_suppl), pp. 2532-2532. |
| ○ | Lee, J. O., Yang, H., Georgescu, M. M., Cristofano, A., Maehama, T., Shi, Y. [...] y Pavletich, N. P. (1999). Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell, 99 (3), pp. 323-334. https://doi.org/10.1016/S0092-8674(00)81663-3 |
| ○ | Liaw, D., Marsh, D. J., Li, J., Dahia, P. L., Wang, S. I., Zheng, Z. [...] y Parsons, R. (1997). Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nature Genetics, 16 (1), pp. 64-67. https://doi.org/10.1038/ng0597-64 |
| ○ | Lindor, N. M., Greene, M. H. y Mayo Familial Cancer Program (1998). The concise handbook of family cancer syndromes. Journal of the National Cancer Institute, 90 (14), pp. 1039-1071. https://doi.org/10.1093/jnci/90.14.1039 |
| ○ | Lloyd, K. M. y Dennis, M. (1963). Cowden´s disease: a possible new symptom complex with multiple system involvement. Annals of Internal Medicine, 58 (1), pp. 136-142. https://doi.org/10.7326/0003-4819-58-1-136 |
| ○ | Lu, Y., Ek, W. E., Whiteman, D., Vaughan, T. L., Spurdle, A. B., Easton, D. F. [...] y Macgregor, S. (2014). Most common ‘sporadic’ cancers have a significant germline genetic component. Human Molecular Genetics, 23 (22), pp. 6112-6118. https://doi.org/10.1093/hmg/ddu312 |
| ○ | Maehama, T. y Dixon, J. E. (1998). The tumor suppressor PTEN/NMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-triphosphate. Journal of Biological Chemistry, 273 (22), pp. 13375-13378. https://doi.org/10.1074/jbc.273.22.13375 |
| ○ | Marsh, D. J. y Zori, R. T. (2002). Genetic insights into familial cancers-update and recent discoveries. Cancer Letters, 181 (2), pp. 125-164. https://doi.org/10.1016/S0304-3835(02)00023-X |
| ○ | McKusick, V. A. (1998). Mendelian Inheritance in Man: a catalog of human genes and genetic disorders. Johns Hopkins University. |
| ○ | Meindl, A., Hellebrand, H., Wiek, C., Erven, V., Wappenschmidt, B., Niederacher, D. [...] y Hanenberg, H. (2010). Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nature Genetics, 42 (5), pp. 410-414. https://doi.org/10.1038/ng.569 |
| ○ | Milne, R. L. y Benítez, J. (2008). Current strategies in the search for low penetrance genes in cancer. Histology and Histopathology, 23 (4), pp. 507-514. |
| ○ | Nagy, R., Sweet, K. y Eng, C. (2004). Highly penetrant hereditary cancer syndromes. Oncogene, 23 (38), pp. 6445-6470. https://doi.org/10.1038/sj.onc.1207714 |
| ○ | Nelen, M. R., Padberg, G. W., Peeters, E. A. J., Lin, A. Y., Helm, B. van den, Frants, R. R. [...] y Eng, C. (1996). Localization of the gene for Cowden disease to chromosome 10q22-23. Nature Genetics, 13 (1), pp. 114-116. https://doi.org/10.1038/ng0596-114 |
| ○ | Ni, Y. y Eng, C. (2012). Vitamin E protects against lipid peroxidation and rescues tumorigenic phenotypes in cowden/cowden-like patient-derived lymphoblast cells with germline SDHx variants. Clinical Cancer Research, 18, pp. 4954-4961. https://doi.org/10.1158/1078-0432.CCR-12-1055 |
| ○ | Nieuwenhuis, M. H., Kets, C. M., Murphy-Ryan, M., Yntema, H. G., Evans, D. G., Colas, C. [...] y Vasen, H. F. A. (2013). Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Familial Cancer, 13 (1), pp. 57-63. https://doi.org/10.1007/s10689-013-9674-3 |
| ○ | Offit, K. (1998). Clinical Cancer Genetics. Risk Counseling & Management. New York: Wiley. |
| ○ | Orloff, M. S., He, X., Peterson, C., Chen, F., Chen, J. L., Mester, J. L. y Eng, C. (2013). Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. The American Journal of Human Genetics, 92 (1), pp. 76-80. https://doi.org/10.1016/j.ajhg.2012.10.021 |
| ○ | Osorio, A., Endt, D., Fernández, F., Eirich, K., Hoya, M. de la, Schmutzler, R. [...] y Benítez, J. (2012). Predominance of pathogenic missense variants in the RAD51C gene occurring in breast and ovarian cancer families. Human Molecular Genetics, 21 (13), pp. 2889-2898. https://doi.org/10.1093/hmg/dds115 |
| ○ | Pascarenco, O. D., Boeriu, A., Mocan, S., Pascarenco, G., Drasoveanu, S., Galeanu, M. y Dobru, D. (2014). Barrett’s esophagus and intestinal metaplasia of gastric cardia: prevalence, clinical, endoscopic and histological features. Journal of Gastrointestinal and Liver Diseases, 23 (1), pp. 19-25. |
| ○ | Pilarski, R., Burt, R., Kohlman, W., Pho, L., Shannon, K. M. y Swisher, E. (2013). Cowden Syndrome and the PTEN Hamartoma Tumor Syndrome: Systematic review and revised diagnostic criteria. Journal of the National Cancer Institute, 105 (21), pp. 1607-1616. https://doi.org/10.1093/jnci/djt277 |
| ○ | Rivera, B., Perea, J., Sánchez, E., Villapun, M., Sánchez-Tomé, E., Mercadillo, F. [...] y Urioste, M. (2014). A novel AXIN2 germline variant associated with attenuated FAP without signs of oligondontia or ectodermal dysplasia. European Journal of Human Genetics, 22 (3), pp. 423-426. https://doi.org/10.1038/ejhg.2013.146 |
| ○ | Roberts, N. J., Jiao, Y., Yu, J., Kopolovich, L., Petersen, G. M., Bondy, M. L. [...] y Klein, A. P. (2012). ATM mutations in patients with hereditary pancreatic cancer. Cancer Discovery, 2, pp. 41-46. https://doi.org/10.1158/2159-8290.CD-11-0194 |
| ○ | Robles-Espinoza, C. D., Harland, M., Ramsay, A. J., Aoude, L. G., Quesada, V., Ding, Z. [...] y Adams, D. J. (2014). POT1 loss-of-function variants predispose to familial melanoma. Nature Genetics, 46 (5), pp. 478-481. https://doi.org/10.1038/ng.2947 |
| ○ | Rosa-Rosa, J. M., Gracia-Aznárez, F. J., Hodges, E., Pita, G., Rooks, M., Xuan, Z. [...] y Benítez, J. (2010). Deep sequencing of target linkage assay-identified regions in familial breast cancer: methods, analysis pipeline and troubleshooting. PLoS One, 5 (4), e9976. https://doi.org/10.1371/journal.pone.0009976 |
| ○ | Salem, O. S. y Steck, W. D. (1983). Cowden´s disease (multiple hamartoma and neoplasia syndrome): A case report and review of the English literature. Journal of the American Academy of Dermatology, 8 (5), pp. 686-696. https://doi.org/10.1016/S0190-9622(83)70081-2 |
| ○ | Schneider, K. (2002). Counselling About Cancer. Strategies for Genetic Counselling (2.ª ed.). New York: Wiley. |
| ○ | Shi, J., Yang, X. R., Ballew, B., Rotunno, M., Calista, D., Fargnoli, M. C. [...] y Landi, M. T. (2014). Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nature Genetics, 46 (5), pp. 482-486. https://doi.org/10.1038/ng.2941 |
| ○ | Snape, K., Ruark, E., Tarpey, P., Renwick, A., Turnbull, C., Seal, S. [...] y Rahman, N. (2012). Predisposition gene identification in common cancers by exome sequencing: insights from familial breast cancer. Breast Cancer Research Treatement, 134 (1), pp. 429-433. https://doi.org/10.1007/s10549-012-2057-x |
| ○ | Sociedad Española de Oncología Médica (SEOM). Las cifras del cáncer en España 2018. [En línea]. Disponible en https://www.seom.org/seomcms/images/stories/recursos/Las_Cifras_del_cancer_en_Espana2018.pdf |
| ○ | Southey, M. C., Park, D. J., Nguyen-Dumont, T., Campbell, I., Thompson, E., Trainer, A. H. [...] y Goldgar, D. E. (2013). COMPLEXO: identifying the missing heritability of breast cancer via next generation collaboration. Breast Cancer Ressearch, 15 (3), pp. 402-402. https://doi.org/10.1186/bcr3434 |
| ○ | Tan, M. H., Mester, J. L., Ngeow, J., Ribicki, L. A., Orloff, M. S. y Eng, C. (2012). Lifetime cancer risks in individuals with germline PTEN mutations. Clinical Cancer Research, 18 (2), pp. 400-407. https://doi.org/10.1158/1078-0432.CCR-11-2283 |
| ○ | Tan, M. H., Mester, J., Peterson, C., Yang, Y., Chen, J. L., Rybicki, L. A [...] y Eng, C. (2011). A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. The American Journal of Human Genetics, 88 (1), pp. 42-56. https://doi.org/10.1016/j.ajhg.2010.11.013 |
| ○ | Thompson, E. R., Doyle, M. A., Ryland, G. L., Rowley, S. M., Choong, D. Y., Tothill, R. W. [...] y Campbell, I. G. (2012). Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genetics, 8 (9), e1002894. https://doi.org/10.1371/journal.pgen.1002894 |
| ○ | Tucker, T., Marra, M. y Friedman, J. M. (2009). Massively parallel sequencing: the next big thing in genetic medicine. The American Journal of Human Genetics, 85 (2), pp. 142-154. https://doi.org/10.1016/j.ajhg.2009.06.022 |
| ○ | Urioste Azcorra, M. (2011). Detección e identificación de síndromes de susceptibilidad al cáncer. En: Bandrés Moya, F. y Urioste Azcorra, M. (eds.). Planteamientos básicos del cáncer hereditario: principales síndromes. Madrid: Fundación Tejerina, pp. 27-46. |
| ○ | Valle, L., Perea, J., Carbonell, P., Fernández, V., Dotor, A. M., Benítez, J. y Urioste, M. (2007). Clinicopathologic and pedigree differences in Amsterdam I-positive hereditary nonpolyposis colorrectal cancer families according to tumor microsatellite instability status. Journal of Clinical Oncology, 25 (7), pp. 781-786. https://doi.org/10.1200/JCO.2006.06.9781 |
| ○ | Vogelstein, B. y Kinzler, K. W. (2002). The Genetic Basis of Human Cancer (2.ª ed.). New York: McGraw-Hill. |
| ○ | Zhang, S. y Yu, D. (2010). PI (3) king apart PTEN´s role in cancer. Clinical Cancer Research, 16, pp. 4325-4330. https://doi.org/10.1158/1078-0432.CCR-09-2990 |
| ○ | Zhuang, Z., Yang, C., Lorenzo, F., Merino, M., Fojo, T., Kebebew, E. [...] y Pacak, K. (2012). Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. The New England Journal of Medicine, 367 (10), pp. 922-930. https://doi.org/10.1056/NEJMoa1205119 |
|