MEDICINA, CIENCIA Y REALIDAD DE LAS ENFERMEDADES RARAS / MEDICINE, SCIENCE AND REALITY OF RARE DISEASES
TERAPIAS AVANZADAS EN ENFERMEDADES RARAS
Cristina Fillat
Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS) / Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER)
ORCID iD: http://orcid.org/0000-0002-0801-3338
cfillat@clinic.cat
Marcela del Río
Universidad Carlos III de Madrid / Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT) / Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) / Instituto de Investigación Sanitaria de la Fundación Jiménez Díaz (IIS-FJD)
ORCID iD: http://orcid.org/0000-0003-2910-7189
mrnechae@ing.uc3m.es
Lucía Martínez-Santamaría
Universidad Carlos III de Madrid / Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT) / Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) / Instituto de Investigación Sanitaria de la Fundación Jiménez Díaz (IIS-FJD)
ORCID iD: http://orcid.org/0000-0002-9637-2364
lmsantam@ing.uc3m.es
Juan A. Bueren
Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT) / Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) / Instituto de Investigación Sanitaria de la Fundación Jiménez Díaz (IIS-FJD)
ORCID iD: http://orcid.org/0000-0002-3228-7013
juan.bueren@ciemat.es
RESUMEN
Las terapias avanzadas comprenden un grupo de medicamentos biológicos basados en la terapia génica, la terapia celular y la ingeniería de tejidos. En este artículo se presentan algunos conceptos básicos, se describen las estrategias más prometedoras y se detallan los resultados de diferentes ensayos clínicos para un grupo significativo de enfermedades, especialmente las enfermedades hereditarias monogénicas. Se hace un mayor hincapié en la terapia génica ex vivo en enfermedades del sistema hematopoyético, al ser el grupo de enfermedades pioneras que además han orientado la investigación en el campo. Destacamos también algunos ejemplos de éxito de terapia génica in vivo y señalamos la contribución de la terapia celular y de la ingeniería de tejidos al grupo de enfermedades raras de la piel. Pretendemos con ello dar una visión de la situación de las terapias avanzadas en enfermedades raras y señalamos algunas de las vías de futuro orientadas al desarrollo de tratamientos más eficaces y seguros.
ADVANCED THERAPIES FOR RARE DISEASES
ABSTRACT
Advanced therapies are a group of medicines for human use based on gene therapy, somatic cell therapy or tissue-engineering. In this paper we present some general basic concepts, describe the most promising strategies and summarize the results of a variety of clinical trials for a significant group of monogenic inherited diseases. Major emphasis is placed on ex vivo gene therapy approaches for diseases of the hematopoietic system, since these corresponded to pioneering treatments that have guided research in this field. We also highlight some examples of successful in vivo gene therapy and point out the contribution of somatic cell therapy and tissue engineering to the group of rare skin diseases. We aim to provide an overview of the state of the art of advanced therapies in rare diseases and highlight some of the cutting-edge technologies under development that can provide more effective and safer treatments in the future.
Recibido: 30-04-2015; Aceptado: 13-05-2016.
Cómo citar este artículo/Citation: Fillat, C.; Río, M. del; Martínez-Santamaría, L. y Bueren, J.A. (2018). Terapias avanzadas en enfermedades raras. Arbor, 194 (789): a467. https://doi.org/10.3989/arbor.2018.789n3009
PALABRAS CLAVE: Terapia génica; terapia celular; ingeniería de tejidos; enfermedades raras.
KEYWORDS: Gene therapy; cell therapy; tissue engineering; rare diseases.
Copyright: © 2018 CSIC. Este es un artículo de acceso abierto distribuido bajo los términos de la licencia de uso y distribución Creative Commons Reconocimiento 4.0 Internacional (CC BY 4.0).
CONTENIDOS
INTRODUCCIÓN Top
Las terapias avanzadas constituyen un grupo de medicamentos de uso humano basados en la terapia génica, la terapia celular o la ingeniería de tejidos. Se trata de estrategias terapéuticas nuevas que abren esperanzas para un gran número de enfermedades raras (ER) que carecen de tratamientos eficaces.
Los medicamentos de terapia avanzada en ER se encuentran en un alto porcentaje de casos en fase de investigación básica y de desarrollo preclínico. En gran medida esto es así puesto que el desarrollo de los mismos va asociado a los avances en los conocimientos de las bases moleculares y fisiopatológicas de la enfermedad. La mayor atención que este grupo de enfermedades, tan heterogéneas y tan poco prevalentes, ha recibido por parte de las instituciones sanitarias en el mundo occidental ha representado un impulso tanto en la investigación básica como en el desarrollo de nuevas terapias, y en especial en terapias avanzadas. Así, para enfermedades de las que se dispone de un buen conocimiento sobre su biología encontramos ensayos clínicos en diferentes fases de desarrollo y empiezan a surgir los primeros medicamentos comercializados, de acuerdo con la legislación existente para medicamentos de terapias avanzadas.
Esta es sin duda un área de presente y futuro y cuyos avances pueden dar respuesta al tratamiento de muchas enfermedades, hoy huérfanas de curación. Este trabajo pretende presentar al lector una visión sobre el estado actual de las terapias avanzadas en enfermedades raras cubriendo aspectos básicos de concepto, diseño y desarrollo, así como presentar algunos ejemplos que de forma exitosa han realizado el largo trayecto que va desde la investigación básica a la práctica clínica.
TERAPIA GÉNICA Top
Conceptos. La terapia génica consiste en un conjunto de estrategias terapéuticas que utilizan un medicamento biológico cuyo principio activo son ácidos nucleicos capaces de modular la expresión de genes endógenos o expresar nuevos genes para prevenir, detener o revertir un proceso patológico. Además del principio activo el medicamento de terapia génica requiere de un vehículo o vector que facilite el acceso del principio activo al interior de la célula diana. Mayoritariamente la terapia génica para enfermedades raras se ha desarrollado alrededor de enfermedades monogénicas recesivas; sin embargo, las posibilidades de una aplicación para enfermedades más complejas está siendo evaluada en estudios preclínicos (Fillat y Altafaj, 2012Fillat, C. y Altafaj, X. (2012). Gene Therapy for Down Syndrome. Progress in Brain Research, 197, pp. 237-247. https://doi.org/10.1016/B978-0-444-54299-1.00012-1.).
La terapia génica, en función de la enfermedad que se vaya a tratar, se desarrolla tomando como base tres principios:
1) Adición génica. Es la estrategia más comúnmente utilizada e implica la adición de una secuencia recombinante de DNA para que exprese un producto génico que está ausente o no es funcional en el paciente. Es una estrategia común para el desarrollo de terapias para enfermedades hereditarias cuyo defecto genético da lugar a una pérdida de función. La adición génica puede darse de manera que el nuevo gen se mantenga de forma episomal en el núcleo de la célula, o bien se integre en el genoma de la célula huésped. Esta integración podrá ser esencialmente aleatoria, o bien podrá tener lugar en una región deseada del genoma, en regiones que no interfieran con la expresión de genes relevantes para la célula, a las que se denomina como zonas seguras del genoma.
2) Supresión génica. En este caso se trata de corregir condiciones patológicas en las que o bien se produce una proteína anómala, o se produce una sobreexpresión patológica de la misma. Estas aproximaciones también se vislumbran para la regulación de ARNs no codificantes, especialmente los miARNs. Así se diseñan secuencias de reconocimiento de miARNs específicos, para que actúen como competidores de los transcritos naturales sobre los que actúan, ejercitando un papel de esponja, o de secuestro de los miARNs.
3) Reparación o edición génica. Esta estrategia se podría considerar la aproximación ideal. El objetivo es sustituir únicamente los cambios nucleotídicos existentes que han dado lugar a un gen disfuncional. Los sistemas desarrollados son similares a los que se utilizan para la inserción de genes en sitios seguros del genoma. Esencialmente consisten en inducir un punto de rotura en una región concreta del ADN, a la vez que se introduce en la célula una región con la secuencia nucleotídica correcta y dos brazos de homología en cada extremo para facilitar que tenga lugar un proceso de recombinación homóloga entre el ADN huésped y el fragmento introducido. Con ello se consigue reparar la región que alberga las mutaciones patogénicas.
Vectores virales: Vectores integrativos. Distinguiremos dos tipos de vectores, los vectores γ-retrovirales y los lentivirales. Ambos comparten algunas características, como que se trata de virus ARN cuyo genoma es fácilmente modificable, y que permiten la expresión en trans de gran parte de sus proteínas para la formación de las partículas virales. Se denominan integrativos ya que el genoma viral se inserta en el ADN cromosómico de la célula infectada y permanece a lo largo de las diferentes divisiones celulares. Estas propiedades los han hecho especialmente atractivos para las estrategias de terapia génica basadas en la manipulación ex vivo de las células. Los vectores γ-retrovirales, y principalmente los derivados del virus de la leucemia murina, fueron los pioneros en entrar en ensayos clínicos de terapia génica. Estos vectores, una vez han infectado la célula, no son capaces de entrar en el núcleo a no ser que la célula se encuentre en división; momento en que la envoltura nuclear se desintegra y el genoma liberado por el vector viral puede tener acceso a los cromosomas celulares y proceder a la integración. La integración de estos vectores tiene lugar principalmente en regiones ricas en genes, y particularmente cerca del inicio de la transcripción. Sin embargo, los vectores lentivirales -los más utilizados derivados del virus VIH- sí son capaces de transportar el genoma al núcleo celular, lo que favorece la transducción de células quiescentes, por lo que este proceso es independiente de la división celular. Como ocurre con los vectores γ-retrovirales, la integración de los vectores lentivirales en el genoma de la célula huésped también transcurre en regiones ricas en genes. No obstante, en este caso no existe preferencia por la integración alrededor del inicio de transcripción (Schröder et al., 2002Schröder, A. R., Shinn, P., Chen, H., Berry, C., Ecker, J. R. y Bushman, F. (2002). HIV-1 integration in the human genome favors active genes and local hotspots. Cell, 110 (4), pp. 521-529. https://doi.org/10.1016/S0092-8674(02)00864-4.). Esto ha hecho que sean considerados vectores más seguros y de mayor aplicabilidad. En estos vectores se realizó también la inactivación de los promotores y secuencias potenciadoras de la expresión, lo que supuso un paso adicional de seguridad. De manera análoga, en los vectores γ-retrovirales también se ha realizado tal inactivación, por lo que en ambos casos se pueden utilizar secuencias reguladoras de la expresión génica más débiles o con actividad específica de linaje celular.
Vectores no integrativos. Destacamos principalmente los vectores derivados de virus adenoasociados (AAV). Se trata de virus ADN, de genoma pequeño y que necesitan de un virus helper, para completar su ciclo de vida. El genoma del AAV puede remplazarse por el gen terapéutico prácticamente en su totalidad, a excepción de las secuencias ITR. Ello permite un margen de clonaje relativamente pequeño, alrededor de las 5kb. Esto es considerado una limitación ya que en ocasiones las secuencias que se quieren introducir exceden este tamaño. Por otro lado, los AAV son vectores altamente atractivos ya que pueden transducir células post-mitóticas, mantienen el genoma de forma episomal en el núcleo celular y hay una variedad significativa de serotipos diferentes de AAV con tropismos de tejido específico. Son vectores de especial interés en estrategias de terapia génica in vivo, en marcha en varios ensayos clínicos. Una de sus principales limitaciones radica en la respuesta inmune que frecuentemente despiertan estos vectores, lo que en algunos casos limita la eficacia del vector.
Estrategias de terapia génica: Ex vivo. La terapia génica ex vivo se basa en la colecta de las células que se quieren modificar del paciente, seguida por la transducción in vitro de estas células con los vectores portadores del gen terapéutico y la posterior reinfusión de las células corregidas al paciente (figura 1). El paradigma de esta estrategia lo constituye la terapia génica ex vivo de células hematopoyéticas, que se describe en detalle más adelante. Esta estrategia se utiliza en la mayor parte de los casos con vectores integrativos que permitan el mantenimiento del gen terapéutico a largo plazo, tales como los vectores γ-retrovirales o lentivirales.
In vivo. La terapia génica in vivo consiste en la administración del vector terapéutico directamente al paciente. La administración puede ser sistémica por vía endovenosa, o local por inyección directa al órgano o tejido de interés (figura 1). Para el tratamiento in vivo de enfermedades raras los principales vectores utilizados son los vectores AAV, cuyos logros se describen más adelante.
|
Figura 1. Modalidades de terapia génica: ex vivo / in vivo. Ex vivo: esquema del protocolo de modificación de células hematopoyéticas mediante vectores gamma-retrovirales / lentivirales. In vivo: esquema de inyección directa de vectores adenoasociados a músculo esquelético
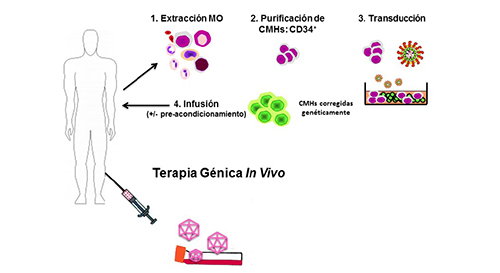
Fuente: Elaboración propia.
[Descargar tamaño completo]
| |
TERAPIA GÉNICA EX VIVO DE ENFERMEDADES RARAS: EL SISTEMA HEMATOPOYÉTICO Top
El protocolo básico seguido en los ensayos clínicos de terapia génica de enfermedades monogénicas del sistema hematopoyético es el que se muestra en la figura 1. En la inmensa mayoría de estos protocolos, las células diana se obtienen a partir de la médula ósea o de aféresis de sangre periférica tras un tratamiento que moviliza las células madre hematopoyéticas (CMH) de la médula ósea. En un paso posterior, las CMHs se purifican mediante sistemas inmunomagnéticos basados en la expresión del marcador CD34, expresado en estas células. Una vez enriquecida la población de CMHs, estas se activan proliferativamente mediante cultivo in vitro durante 24 o 28 horas, seguido de uno o varios ciclos de transducción con los vectores γ-retrovirales o lentivirales portadores de los genes terapéuticos durante un máximo de 2 o 3 días adicionales. Por último, las células CD34+ sometidas al proceso de corrección genética se infunden en el paciente por vía endovenosa.
En las patologías en las que las células corregidas desarrollan fuerte ventaja proliferativa respecto a las células no corregidas, como es el caso de la inmunodeficiencia X1-SCID, las células se pueden infundir sin necesidad de pre-acondicionar a los pacientes con quimioterapia. Por el contrario, si las células corregidas no desarrollan una ventaja proliferativa, el paciente recibe un acondicionamiento quimioterápico para facilitar el injerto de las células transducidas.
Imunodeficiencia severa combinada por déficit de ADA. Entre los síndromes hematopoyéticos que primero se consideraron para su tratamiento génico destaca la inmunodeficiencia severa combinada asociada a mutaciones en el gen Adenosina Deaminasa (ADA-SCID). La ausencia de ADA implica la acumulación del sustrato desoxiadenosina trifosfato en la célula, lo cual resulta particularmente tóxico en los linfocitos T. Esta inmunodeficiencia fue una de las primeras patologías que se trataron mediante terapia génica con vectores γ-retrovirales, primero en los National Institutes of Health (NIH) de Estados Unidos (Blaese et al., 1995Blaese, R. M., Culver, K. W., Miller, A. D., Carter, C. S., Fleisher, T., Clerici, M., Shearer, G. [...] y Anderson, W. F. (1995). T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science, 270 (5235), pp. 475-480. https://doi.org/10.1126/science.270.5235.475.; Kohn et al., 1998Kohn, D. B., Hershfield, M. S., Carbonaro, D., Shigeoka, A., Brooks, J., Smogorzewska, E. M. [...] y Parkman, R. (1998). T lymphocytes with a normal ADA gene accumulate after transplantation of transduced autologous umbilical cord blood CD34+ cells in ADA-deficient SCID neonates. Nature Medicine, 4 (7), pp. 775-780. https://doi.org/10.1038/nm0798-775.), y luego en el Hospital San Raffaelle de Milán (Aiuti et al., 2002Aiuti, A., Slavin, S., Aker, M., Ficara, F., Deola, S., Mortellaro, A. [...] y Bordignon, C. (2002). Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science, 296 (5577), pp. 2410-2413. https://doi.org/10.1126/science.1070104.; Bordignon et al., 1993Bordignon, C., Mavilio, F., Ferrari, G., Servida, P., Ugazio, A. G., Notarangelo, L. D. [...] y Gillio, A. P. (1993). Transfer of the ADA gene into bone marrow cells and peripheral blood lymphocytes for the treatment of patients affected by ADA-deficient SCID. Human Gene Therapy, 4 (4), pp. 513-520. https://doi.org/10.1089/hum.1993.4.4-513.). Las alternativas que se consideraron para el tratamiento genético de esta enfermedad tenían por objeto la transferencia del gen ADA en los linfocitos T de los pacientes o en las CMHs. Tras la infusión de las células corregidas en los pacientes, se observó la presencia de bajas proporciones de células corregidas en sangre y modestas evidencias de beneficio clínico.
Los mejores resultados se obtuvieron posteriormente por parte del equipo del Dr. A. Aiuti en Milán, quien introdujo modificaciones sustanciales en los ensayos clínicos anteriores. Como paso previo a la infusión de las células CD34+ transducidas, este investigador retiró la administración de la proteína ADA recombinante a los pacientes. Con ello se pretendía forzar la ventaja proliferativa de las células corregidas frente a las no corregidas. Por otra parte, para facilitar el injerto de las células infundidas, los pacientes se acondicionaron con un tratamiento submieloablativo de busulfán (Aiuti et al., 2002Aiuti, A., Slavin, S., Aker, M., Ficara, F., Deola, S., Mortellaro, A. [...] y Bordignon, C. (2002). Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science, 296 (5577), pp. 2410-2413. https://doi.org/10.1126/science.1070104.; Aiuti y Roncarolo, 2009Aiuti, A. y Roncarolo, M. G. (2009). Ten years of gene therapy for primary immune deficiencies. Hematology. American Society of Hematology Education Program Book, 2009 (1), pp. 682-689.).
Desde el punto de vista clínico, este protocolo mostró beneficios terapéuticos incuestionables en los diez pacientes a quienes se les aplicó en el primer ensayo clínico. A los diez años de tratamiento, ocho de los pacientes mantenían la función de los linfocitos T sin tratamiento con la proteína ADA recombinante. Es de destacar que en este protocolo todos los pacientes tratados estaban vivos, y ninguno de los pacientes manifestó efectos adversos severos como consecuencia de fenómenos de mutagénesis insercional. A día de hoy se han tratado más de 40 pacientes con este protocolo, siendo la supervivencia del 100% y la eficacia terapéutica del 75%. Este protocolo constituye uno de los ejemplos más representativos de la eficacia y seguridad asociada a procedimientos de terapia génica ex vivo. En virtud de los resultados obtenidos, el medicamento consistente en células CD34+ transducidas con el vector γ-retroviral utilizado en este ensayo recibió en 2016 la autorización de la Agencia Europea del Medicamento para ser registrado como medicamento de uso clínico en Europa, bajo la denominación de Strimvelis (Aiuti, Roncarolo y Naldini, 2017Aiuti, A., Roncarolo, M. G. y Naldini, L. (2017). Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: paving the road for the next generation of advanced therapy medicinal products. EMBO Molecular Medicine, 9 (6), e201707573. https://doi.org/10.15252/emmm.201707573.).
Inmunodeficiencia severa combinada X1-SCID. La inmunodeficiencia X1-SCID representa aproximadamente la mitad de todas las inmunodeficiencias severas combinadas y está asociada a un defecto en la cadena gc, una proteína que forma parte de numerosos receptores de interleuquinas.
La terapia génica de estos pacientes se inició en el Hospital Necker de París, por parte de los Drs. Fischer y Cavazzana, mediante la transferencia del gen terapéutico a células CD34+, utilizando vectores γ-retrovirales. En este caso, la infusión se realizó sobre pacientes que no habían recibido acondicionamiento alguno. En los dos primeros pacientes que se trataron se consiguió restaurar tanto el número como la función de las células B, T y NK (Cavazzana-Calvo et al., 2000Cavazzana-Calvo, M., Hacein-Bey, S., Saint Basile, G. de, Gross, F., Yvon, E., Nusbaum, P. [...] y Fischer, A. (2000). Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science, 288 (5466), pp. 669-672. https://doi.org/10.1126/science.288.5466.669.). Estos resultados se confirmaron en cinco nuevos pacientes tratados en el mismo centro (Hacein-Bey-Abina et al., 2002Hacein-Bey-Abina, S., Deist, F. Le, Carlier, F., Bouneaud, C., Hue, C., Villartay, J. P. de [...] y Cavazzana-Calvo, M. (2002). Sustained correction of X-Linked severe combined immunodeficiency by ex vivo gene therapy. The New England Journal of Medicine, 346 (16), pp. 1185-1193. https://doi.org/10.1056/NEJMoa012616.) y en otros cuatro pacientes tratados por B. Gaspar y A. Thrasher en Londres (Gaspar et al., 2004Gaspar, H. B., Parsley, K. L., Howe, S., King, D., Gilmour, K. C., Sinclair, J. [...] y Thrasher, A. J. (2004). Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. The Lancet, 364 (9452), pp. 2181-2187. https://doi.org/10.1016/S0140-6736(04)17590-9.). A partir del segundo año de tratamiento, no obstante, seis de los veinte pacientes X1-SCID que fueron infundidos con células transducidas con los vectores γ-retrovirales desarrollaron leucemias linfocíticas, si bien cinco de ellos respondieron satisfactoriamente al tratamiento antitumoral.
La inmunodeficiencia X1-SCID y los avances en la seguridad de la terapia génica. En los pacientes X1-SCID que desarrollaron leucemia linfocítica se observó que una gran proporción de las inserciones del vector terapéutico tuvieron lugar en regiones próximas o dentro de genes de las células diana. De hecho, se observó que con alta frecuencia la inserción del vector terapéutico ocurría junto al oncogén LMO2, implicado en leucemias linfocíticas (Hacein-Bey-Abina et al., 2003Hacein-Bey-Abina, S., Saint Basile, G. de y Cavazzana-Calvo, M. (2003). Gene therapy of X-linked severe combined immunodeficiency. Methods in Molecular Biology, 215, pp. 247-259.). Con objeto de minimizar el riesgo de transactivar oncogenes por la inserción de vectores terapéuticos, se desarrollaron nuevos vectores γ-retrovirales en los cuales las regiones LTR se habían inactivado a semejanza de los realizados en los vectores lentivirales autoinactivados. Con este vector ninguno de los más de diez pacientes tratados ha desarrollado reacciones adversas asociadas a la inserción del vector (Hecein-Bey-Abina et al., 2014Hacein-Bey-Abina, S., Pai, S. Y., Gaspar, H. B., Armant, M., Berry, C. C., Blanche, S. [...] y Thrasher, A. J. (2014). A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. The New England Journal of Medicine, 371 (15), pp. 1407-1417. https://doi.org/10.1056/NEJMoa1404588.).
Granulomatosis crónica. La granulomatosis crónica constituye otra inmunodeficiencia que ha sido tratada mediante terapia génica. Esta enfermedad se caracteriza por una respuesta ineficaz de las células fagocíticas para generar anión superóxido, lo que se manifiesta mediante un síndrome recurrente de infecciones y formación de granulomas.
Los estudios clínicos realizados por el Dr. Manuel Grez en Frankfurt sobre pacientes con mutaciones en el gen pg91 phox y basados en la infusión de células CD34+ transducidas con vectores γ-retrovirales en dos pacientes adultos que recibieron acondicionamiento mieloablativo también mostraron mejorías clínicas, pues los pacientes tratados en estos estudios generaron células fagocíticas con actividad NADPH funcional (Ott et al., 2006Ott, M. G., Schmidt, M., Schwarzwaelder, K., Stein, S., Siler, U., Koehl, U. [...] y Grez, M. (2006). Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nature Medicine, 12 (4), pp. 401-409. https://doi.org/10.1038/nm1393.). A pesar de ello, y tal como ocurrió en el caso de la inmunodeficiencia X1-SCID, también se observaron fenómenos de oncogénesis insercional que promovieron una reconstitución oligoclonal de los pacientes a partir de los clones en los que se había producido la transactivación de oncogenes tales como MDS-EVI1 (Stein et al., 2010Stein, S., Ott, M. G., Schultze-Strasser, S., Jauch, A., Burwinkel, B., Kinner, A. [...] y Grez, M. (2010). Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nature Medicine, 16 (2), pp. 198-204. https://doi.org/10.1038/nm.2088.). Como consecuencia de ello, estos ensayos clínicos se detuvieron hasta el desarrollo de nuevos vectores. Recientemente se ha puesto en marcha un ensayo clínico en donde el gen GP91 está insertado en un vector lentiviral autoinactivado, que está demostrando evidencias de injerto estable con células que han recuperado la función fagocítica, con clara mejoría clínica y ausencia de genotoxicidad.
β-Talasemia. En el año 2010 la Dra. Marina Cavazzana publicó los resultados del primer paciente con b-talasemia que fue tratado con células CD34+ que se transdujeron con un vector lentiviral que confería expresión específica del gen de la globina-b en células de la serie eritroide. Tras un acondicionamiento mieloablativo, los pacientes se infundieron con las células transducidas. Progresivamente a lo largo del tiempo se observó el incremento de la población de células eritroides portadoras del gen terapéutico (Cavazzana-Calvo et al., 2010Cavazzana-Calvo, M., Payen, E., Negre, O., Wang, G., Hehir, K., Fusil, F. [...] y Leboulch, P. (2010). Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature, 467 (7313), pp. 318-322.).
Durante un tiempo, se observó la predominancia de células que provenían de un clon celular en donde el vector lentiviral se había integrado en un microARN (Let-7), produciendo su inactivación y con ello la sobre-expresión de uno de sus genes diana (HMGA2), implicado en crecimiento celular. Afortunadamente, tras un periodo de predominancia clonal benigna, la hematopoyesis del paciente retornó a un patrón policlonal, disminuyendo los riesgos de progresión maligna. Nuevos pacientes han sido tratados con este tipo de vectores tanto en Europa como en Estados Unidos, sin que por el momento se hayan indicado efectos adversos severos. Más recientemente la Dra. Giuliana Ferrari, del Hospital San Raffaele, también ha demostrado beneficio clínico en tres de los cuatro pacientes tratados con un nuevo vector lentiviral.
Síndrome de Wiskott-Aldrich. Se trata de un síndrome asociado a inmunodeficiencia y trombocitopenia. En el año 2010 el equipo del Dr. C. Klein publicó los resultados de los dos primeros pacientes tratados con un vector γ-retroviral portador del gen de Wiskott-Aldrich. Estos pacientes mostraron una mejoría clínica evidente, resolviéndose los problemas hemorrágicos, eccema, autoinmunidad y predisposición a infecciones severas (Boztug et al., 2010Boztug, K., Schmidt, M., Schwarzer, A., Banerjee, P. P., Díez, I. A., Dewey, R. A. [...] y Klein, C. (2010). Stem-cell gene therapy for the Wiskott-Aldrich syndrome. The New England Journal of Medicine, 363 (20), pp. 1918-1927. https://doi.org/10.1056/NEJMoa1003548.). El beneficio terapéutico observado en este ensayo se confirmó en un estudio posterior (Braun et al., 2014Braun, C. J., Boztug, K., Paruzynski, A., Witzel, M., Schwarzer, A., Rothe, M. [...] y Klein, C. (2014). Gene therapy for Wiskott-Aldrich syndrome--long-term efficacy and genotoxicity. Science Transational. Medicine, 6 (227), 227ra33. https://doi.org/10.1126/scitranslmed.3007280.). No obstante, como ocurrió en el tratamiento de pacientes X1-SCID con vectores γ-retrovirales, progresivamente se observó la aparición de leucemias mieloides y linfoides en siete de los diez pacientes tratados como consecuencia de fenómenos de genotoxicidad.
En virtud de estos fenómenos de oncogénesis insercional, el Dr. A. Aiuti inició un ensayo clínico con vectores lentivirales en pacientes con Wiskott-Aldrich. En la actualidad se han publicado los resultados obtenidos en tres pacientes, en los que además de demostrar la recuperación en el recuento de plaquetas y el rescate de la inmunodeficiencia, también se demuestra que el patrón de reconstitución hematopoyética de los pacientes fue policlonal, sin que por el momento se hayan generado efectos adversos en ninguno de los pacientes tratados (Aiuti et al., 2013Aiuti, A., Biasco, L., Scaramuzza, S., Ferrua, F., Cicalese, M. P., Baricordi, L. [...] y Naldini, L. (2013). Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science, 341 (6148), 1233151. https://doi.org/10.1126/science.1233151.).
Adrenoleucodistrofia y leucodistrofia metacromática. La transducción de células madre hematopoyéticas se ha utilizado no solo para la corrección de enfermedades de este sistema, sino también para el tratamiento de otras patologías, tales como diferentes leucodistrofias. En estos casos, el objetivo clínico perseguido es el de hacer llegar la proteína deficitaria en el paciente a los tejidos críticos, tal como es el sistema nervioso, en estas patologías. Puesto que estas dos leucodistrofias se habían tratado anteriormente mediante trasplante alogénico de CMHs, se pensó que la infusión de CMH autólogas que sobre-expresaran la proteína deficitaria en el paciente podría también restaurar o al menos detener la progresión de la enfermedad.
La X-adrenoleucodistrofia cursa con una desmielinización severa en el cerebro que se produce como consecuencia de la deficiencia en la proteína ALD en los oligodendrocitos y microglía. En el ensayo clínico realizado por la Dra. N. Cartier las células CMHs se transdujeron con vectores lentivirales portadores del gen de la ALD y posteriormente se reinfundieron en los pacientes tras un acondicionamiento mieloablativo para facilitar la reconstitución a partir de las CMHs transducidas. Los resultados que se presentaron en dos pacientes demostraron por primera vez que era posible detener el proceso de desmielinización en estos pacientes, de manera análoga a como se había mostrado en pacientes sometidos a trasplante alogénico de CMHs, pero sin las complicaciones clínicas asociadas a un trasplante alogénico (Cartier et al., 2009Cartier, N., Hacein-Bey-Abina, S., Bartholomae, C. C., Veres, G., Schmidt, M., Kutschera, I. [...] y Aubourg, P. (2009). Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science, 326 (5954), pp. 818-823. https://doi.org/10.1126/science.1171242.).
Posteriormente, los Drs. A. Biffi y L. Naldini también obtuvieron resultados positivos en un ensayo en tres pacientes asintomáticos con leucodstrofia metacromática, originada por una deficiencia en el gen de la arilsulfatasa A (ARSA). Como en el caso de la ALD, en los tres pacientes tratados en este ensayo se observó un detenimiento en el progreso de la enfermedad, sin que se manifestaran efectos adversos colaterales (Biffi et al., 2013Biffi, A., Montini, E., Lorioli, L., Cesani, M., Fumagalli, F., Plati, T. [...] y Naldini, L. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science, 341 (6148), 1233158. https://doi.org/10.1126/science.1233158.).
Próximos ensayos clínicos de enfermedades monogénicas del sistema hematopoyético. La seguridad y eficacia clínica observada en los ensayos clínicos de terapia génica ha promovido el desarrollo de nuevos vectores que permitan la puesta en marcha de nuevos ensayos clínicos para el tratamiento génico de otros síndromes que afectan a células del sistema hematopoyético. Así, la Unidad 710 del CIBERER (CIEMAT/IIS Fundación Jiménez Díaz) ha desarrollado nuevos vectores lentivirales para el tratamiento de tres graves patologías que cursan con anemia aplásica (anemia de Fanconi), anemia macrocítica (deficiencia en piruvato quinasa) e inmunodeficiencia (deficiencia de adhesión leucocitaria de tipo I). La anemia de Fanconi (AF) se caracteriza principalmente por fallo de médula ósea y predisposición a cáncer. Los ensayos clínicos realizados en Cincinnati por el equipo del Dr. D. Williams con vectores γ-retrovirales no generaron beneficios terapéuticos en los pacientes (Kelly et al., 2007Kelly, P. F., Radtke, S., Kalle, C. von, Balcik, B., Bohn, K., Mueller, R. [...] y Williams, D. A. (2007). Stem cell collection and gene transfer in Fanconi anemia. Molecular Therapy, 15 (1), pp. 211-219. https://doi.org/10.1038/sj.mt.6300033.). En virtud de ello, nuestro equipo de investigación ha venido trabajando en el desarrollo de nuevos vectores lentivirales, más eficaces y seguros, que abren nuevas expectativas para el tratamiento de pacientes con anemia de Fanconi del subtipo A, que afecta al 80% de los pacientes españoles con AF (Adair et al., 2016Adair, J. E., Sevilla, J., Díaz de Heredia, C., Becker, P. S., Kiem, H-P. y Bueren, J. A. (2016). Lessons Learned from Two Decades of Clinical Trial Experience in Gene Therapy for Fanconi Anemia. Current Gene Therapy, 16 (5), pp. 338-348. https://doi.org/10.2174/1566523217666170119113029.; Gonzalez-Murillo et al., 2010González-Murillo, A., Lozano, M. L., Álvarez, L., Jacome, A., Almarza, E., Navarro, S. [...] y Río, P. (2010). Development of lentiviral vectors with optimized transcriptional activity for the gene therapy of patients with Fanconi anemia. Human Gene Therapy, 21 (5), pp. 623-630. https://doi.org/10.1089/hum.2009.141.; Tolar et al., 2012Tolar, J., Becker, P. S., Clapp, D. W., Hanenberg, H., Heredia, C. D. de, Kiem, H. P. [...] y Bueren, J. (2012). Gene therapy for Fanconi anemia: one step closer to the clinic. Human Gene Therapy, 23 (2), pp. 141-144. https://doi.org/10.1089/hum.2011.237.). En virtud de los estudios preclínicos realizados, la Comisión Europea y la Food and Drug Administration (FDA) americana designaron el vector desarrollado por nuestro equipo como un nuevo medicamento huérfano para el tratamiento de pacientes con anemia de Fanconi. Asimismo, la Agencia Española del Medicamento aprobó en 2013 el protocolo de terapia génica en España, el cual ha recibido financiación del Ministerio de Sanidad y de la Comisión Europea para su puesta en marcha en España y en otros países europeos. Resultados muy recientes han mostrado que la transducción de CMHs de pacientes AF con nuestro vector lentiviral son capaces de reconstituir el sistema hematopoyético de ratones inmunodeficientes (Río et al., 2017Río, P., Navarro, S., Guenechea, G., Sánchez-Domínguez, R., Lamana, M. L., Yañez, R. [...] y Bueren, J. (2017). Engraftment and in vivo proliferation advantage of gene corrected mobilized CD34+ cells from Fanconi anemia patients. Blood, 130 (13), pp. 1535-1542. https://doi.org/10.1182/blood-2017-03-774174.), lo que abre nuevas esperanzas a que la terapia génica sea un tratamiento eficaz y poco tóxico para pacientes con anemia de Fanconi.
En el caso de la anemia producida por déficit en piruvato quinasa eritrocitaria, los estudios preclínicos ya realizados con este vector muestran no solo su eficacia terapéutica sino también su seguridad (García-Gómez et al., 2016García-Gómez, M., Calabria, A., García-Bravo, M., Benedicenti, F., Kosinski, P., López-Manzaneda, S. [...] y Segovia, J. C. (2016). Safe and Efficient Gene Therapy for Pyruvate Kinase Deficiency. Molecular Therapy, 24 (7), pp. 1187-1198. https://doi.org/10.1038/mt.2016.87.; Meza et al., 2009Meza, N. W., Alonso-Ferrero, M. E., Navarro, S., Quintana-Bustamante, O., Valeri, A., García-Gómez, M. [...] y Segovia, J. C. (2009). Rescue of pyruvate kinase deficiency in mice by gene therapy using the human isoenzyme. Molecular Therapy, 17 (12), pp. 2000-2009. https://doi.org/10.1038/mt.2009.200.), por lo que confiamos en dar comienzo a los estudios clínicos en los próximos años. En esta línea de trabajo, la Agencia Europea del Medicamento y la FDA han designado también al vector lentiviral desarrollado por esa Unidad CIBERER como medicamento huérfano.
Una tercera enfermedad en la que venimos trabajando en esta Unidad CIBERER es la inmunodeficiencia conocida como deficiencia de adhesión leucocitaria de tipo I (LAD-1). Como en los casos anteriores los estudios preclínicos realizados con el vector lentiviral construido por nuestro laboratorio han mostrado evidencias de eficacia terapéutica (León-Rico et al., 2016León-Rico, D., Aldea, M., Sánchez-Baltasar, R., Mesa-Núñez, C., Record, J., Burns, S. O. [...] y Almarza, A. (2016). Lentiviral vector mediated correction of a mouse model of Leukocyte Adhesion Deficiency Type I. Human Gene Therapy, 27 (9), pp. 668-678. https://doi.org/10.1089/hum.2016.016.), lo que ha facilitado también la designación de medicamento huérfano por las agencias europea y americana. Confiamos en que el uso de este vector lentiviral portador del gen CD18, deficiente en los pacientes con LAD-1, nos permita próximamente iniciar el tratamiento de pacientes con esta grave inmunodeficiencia.
TERAPIA GÉNICA IN VIVO DE ENFERMEDADES RARAS: TEJIDOS DIANA Top
La retina. Probablemente los ensayos clínicos desarrollados hasta ahora para enfermedades de degeneración de la retina se pueden considerar uno de los mayores logros de terapia génica in vivo con vectores adenoasociados. La amaurosis congénita de Leber (ACL) fue la primera enfermedad de este grupo que entró en protocolos de terapia génica. Se trata de una enfermedad genética para la que se han descrito hasta 14 genes cuyas mutaciones dan lugar a diferentes subtipos de la enfermedad. Para el subtipo que cursa con mutaciones en el gen RPE65 (retinal pigment epithelium-specific 65 KDa protein) tres grupos, uno en Reino Unido y dos en Estados Unidos, iniciaron ensayos de terapia génica en 2007 (Bainbridge et al., 2008Bainbridge, J. W., Smith, A. J., Barker, S. S, Robbie, S., Henderson, R., Balaggan, K. [...] y Ali, R. R. (2008). Effect of gene therapy on visual function in Leber’s congenital amaurosis. The New England Journal of Medicine, 358 (21), pp. 2231-2239. https://doi.org/10.1056/NEJMoa0802268.; Hauswirth et al., 2008Hauswirth, W. W., Aleman, T. S., Kaushal, S., Cideciyan, A. V., Schwartz, S. B., Wang, L. [...] y Jacobson, S. G. (2008). Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Human Gene Therapy, 19 (10), pp. 979-990. https://doi.org/10.1089/hum.2008.107.; Maguire et al., 2008Maguire, A. M., Simonelli, F., Pierce, E. A., Pugh Jr., E. N, Mingozzi, F., Bennicelli, J. [...] y Bennett, J. (2008). Safety and efficacy of gene transfer for Leber’s congenital amaurosis. The New England Journal Medicine, 358 (21), pp. 2240-2248. https://doi.org/10.1056/NEJMoa0802315.). Estos estudios demostraron la seguridad del tratamiento, con una respuesta inmune frente al virus o al transgén mínima, probablemente por ser el ojo un lugar inmunoprivilegiado. Se han publicado respuestas parciales en algunas de las funciones estudiadas y las mejoras se han mantenido a lo largo de varios años. La readministración en el segundo ojo de un mismo paciente no ha dado lugar a ningún efecto adverso y en cambio sí ha permitido mejorar la visión (Bennett et al., 2012Bennett, J., Ashtari, M., Wellman, J., Marshall, K. A., Cyckowski, L. L., Chung, D. C. [...] y Maguire, A. M. (2012). AAV2 gene therapy readministration in three adults with congenital blindness. Science Translational Medicine, 4 (120), 120ra15. https://doi.org/10.1126/scitranslmed.3002865.). La agencia americana FDA ha aprobado la terapia génica para ACL, que sería el primer tratamiento de terapia génica para una enfermedad genética hereditaria aprobado en Estados Unidos.
El grupo de enfermedades con alteraciones en la visión es muy amplio y se están iniciando ensayos clínicos en nuevas enfermedades, como la degeneración macular asociada a la edad (NCT01024998), el síndrome de Usher tipo B (NCT01505062) o la enfermedad de Stargardt (NCT01367444), esta última mediante el uso de vectores lentivirales.
El hígado. Entre las enfermedades raras cuyo órgano diana para la expresión del transgén es el hígado, destacamos dos. En primer lugar, la hemofilia B, una enfermedad causada por mutaciones en el factor IX de la coagulación de la sangre, para la que se realizaron ensayos clínicos tras la administración endovenosa de un vector adenoasociado serotipo 2 que expresaba el factor IX. En uno de los sujetos tratados se observaron signos de eficacia con una dosis de factor circulante entre el 10 y el 12 por ciento de un individuo normal. Sin embargo, el factor persistió únicamente a lo largo de diez semanas como consecuencia de la destrucción de las células hepáticas transducidas por linfocitos T activados frente a péptidos de la cápside del AAV. Estos datos, que no se habían visto antes en modelos animales, demuestran las diferencias existentes entre especies con relación a la presentación antigénica y reactividad de las células T (Mingozzi y High, 2011Mingozzi, F. y High, K. A. (2011). Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nature Reviews Genetics, 12 (5), pp. 341-355. https://doi.org/10.1038/nrg2988.). Posteriormente, con virus AAV serotipo 8 más optimizados se desarrollaron nuevos ensayos clínicos para hemofilia B. Se utilizaron dosis bajas del vector y aun así pudieron observarse valores de entre el 3 y el 11 por ciento del factor IX circulante, y los pequeños efectos adversos observados pudieron ser controlados mediante tratamiento puntual de glucocorticodes.
Otra de las enfermedades para las que el hígado es el órgano diana es la porfiria aguda intermitente, cuyas alteraciones son debidas a mutaciones en el gen de la porfobilinógeno deaminasa. Liderado por un grupo español del Centro de Investigación Médica Aplicada (CIMA) de Navarra, se ha llevado a cabo un ensayo clínico fase 1 con un vector AAV, serotipo 5 que expresa de forma hepatoespecífica la porfobilinógeno deaminasa (NCT02082860), mostrando seguridad del tratamiento, si bien a las dosis utilizadas no se observó una corrección del defecto metabólico.
El músculo esquelético. Este tejido, en parte como consecuencia de su facilidad de acceso, es altamente atractivo para la aplicación de terapias, no solo de enfermedades musculares sino también de enfermedades cuya proteína sea secretada fácilmente. Destacamos el tratamiento de la deficiencia familiar de lipoproteinlipasa, un trastorno genético debido a mutaciones en el gen de la lipoproteinlipasa que conlleva defectos en la degradación de la grasa con niveles altos de triglicéridos y pancreatitis recurrentes. El tratamiento intramuscular con AAV serotipo 1 que expresa el gen de la lipoprotein lipasa consigue reducir el número de episodios de pancreatitis. El producto AAV2/1-LPL, conocido con el nombre de Glybera, fue aprobado para su comercialización por parte de la Comisión Europea en noviembre de 2012, convirtiéndose así en el primer medicamento de terapia génica en el mercado.
Próximos ensayos clínicos de enfermedades monogénicas mediante estrategias in vivo. Entre las enfermedades para las que se prevé el inicio de ensayos clínicos, destacamos la mucopolisacaridosis tipo III A o síndrome de Sanfilippo A. Es una enfermedad metabólica debida a mutaciones en el gen de la sulfamidasa que cursa con importante alteraciones somáticas y neurológicas. A raíz de los estudios preclínicos del grupo de la Dra. Bosch en el CBATEG Universidad Autónoma de Barcelona (Haurigot et al., 2013Haurigot, V., Marcó, S., Ribera, A., García, M., Ruzo, A., Villacampa, P. [...] y Bosch, F. (2013). Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. The Journal of Clinical Investigation, 123 (8), pp. 3254-3271. https://doi.org/10.1172/JCI66778.), la Comisión Europea ha designado el vector adenoasociado que contiene el gen de la sulfamidasa humana como medicamento huérfano. En colaboración con Laboratorios Esteve el proyecto de Sanfilippo A ha entrado en fase clínica recientemente.
Como resultado del trabajo liderado por grupos españoles, también han recibido la designación de medicamentos huérfanos los vectores adenoasociados que contienen el gen de la timina fosoforilasa para el tratamiento de la enfermedad de MNGIE, tomando como base los estudios preclínicos de los Dres. Martí y Barquinero (Torres-Torronteras et al., 2014Torres-Torronteras, J., Viscomi, C., Cabrera-Pérez, R., Cámara, Y., Meo, I. di, Barquinero, J. [...] y Martí, R. (2014). Gene therapy using a liver-targeted AAV vector restores nucleoside and nucleotide homeostasis in a murine model of MNGIE. Molecular Therapy, 22 (5), pp. 901-907. https://doi.org/10.1038/mt.2014.6.) y el vector adenoasociado portador del gen de la alanina glioxilato aminotransferasa para el tratamiento de la hyperoxaluria primaria tipo 1, liderado por el Dr. Salido (Salido et al., 2011Salido, E., Rodríguez-Pena, M., Santana, A., Beattie, S. G., Petry, H. y Torres, A. (2011). Phenotypic correction of a mouse model for primary hyperoxaluria with adeno-associated virus gene transfer. Molecular Therapy, 19 (5), pp. 870-875. https://doi.org/10.1038/mt.2010.270.).
INGENIERÍA DE TEJIDOS Top
Dentro del ámbito de la medicina regenerativa, la ingeniería de tejidos (IT) es un campo emergente y muy dinámico de carácter multidisciplinar que combina la investigación básica y la clínica con el objetivo de desarrollar sustitutos biológicos para reemplazar in vivo tejidos y órganos dañados a causa de enfermedades, lesiones y anomalías congénitas, además de estimular la capacidad regenerativa intrínseca del organismo.
Los tres elementos clave de la IT son las células, la matriz o andamiaje empleado y los morfógenos o factores de crecimiento (Atala, 2007Atala, A. (2007). Engineering tissues, organs and cells. Journal of Tissue Engineering and Regenerative. Medicine, 1 (2), pp. 83-96. https://doi.org/10.1002/term.18.), que han de combinarse adecuadamente para reproducir una estructura tridimensional que sea morfológica y funcionalmente similar al tejido u órgano que desee reemplazar y en la cual sea posible el mantenimiento y la diferenciación de los diversos tipos celulares hacia el fenotipo correcto. La dificultad de recrear andamiajes tridimensionales complejos capaces de albergar distintos tipos celulares ha llevado al desarrollo de técnicas de re-celularización de “esqueletos” de órganos previamente descelularizados como es el caso de la tráquea o de órganos complejos como el corazón y el hígado (Song y Ott, 2011Song, J. J. y Ott, H. C. (2011). Organ engineering based on decellularized matrix scaffolds. Trends in Molecular Medicine, 17 (8), pp. 424-432. https://doi.org/10.1016/j.molmed.2011.03.005.). Otros abordajes novedosos que están permitiendo el desarrollo de estructuras tridimensionales complejas son las técnicas de bioprinting, que se basan en las tecnologías de impresión en 3D, haciendo posible la generación de estructuras tisulares tridimensionales y dando lugar a mini-órganos completos (Bajaj, Schweller, Khademhosseini, West y Bashir, 2014Bajaj, P., Schweller, R. M., Khademhosseini, A., West, J. L. y Bashir, R. (2014). 3D biofabrication strategies for tissue engineering and regenerative medicine. Annual Review of Biomedical Engineering, 16, pp. 247-276. https://doi.org/10.1146/annurev-bioeng-071813-105155.).
La piel como sistema modelo. La piel, el órgano externo que nos protege del medio ambiente, está formada por dos compartimentos, la epidermis y la dermis. La epidermis es un epitelio estratificado y queratinizado, dotado de una gran capacidad regenerativa esencial en el que el principal tipo celular es el queratinocito. Por debajo se encuentra la dermis, un tejido conectivo especializado, poblado fundamentalmente por fibroblastos, células de origen mesenquimal productoras de matriz extracelular. La regeneración continua de la epidermis tiene lugar a expensas de la proliferación de una subpoblación del estrato basal, las células madre de la epidermis (CMEs). La comprensión de la biología fundamental de las CMEs y de los mecanismos que rigen su multipotencialidad es clave para el éxito de estrategias terapéuticas basadas en IT, terapia génica o terapia celular en el campo de la medicina regenerativa cutánea.
La IT cutánea surgió motivada principalmente por la necesidad crítica de dar cobertura permanente a grandes quemados, y posteriormente se ha empleado también en el tratamiento de úlceras crónicas. La posibilidad de fabricar in vitro grandes cantidades de epitelio cultivado a partir de una pequeña muestra de piel del paciente revolucionó la terapéutica de los grandes quemados. Si bien la piel fue el primer órgano producido mediante técnicas de IT, el campo de la IT cutánea ha evolucionado vertiginosamente en las últimas décadas, permitiendo importantes avances en el campo de la regeneración cutánea. En sus inicios, se emplearon cultivos 2D (una monocapa de queratinocitos) y posteriormente se han ido desarrollando nuevos sistemas de cultivo celular organotípicos basados en el uso de biomateriales que tratan de mimetizar la dermis humana. Como resultado, una amplia variedad de sustitutos cutáneos bioingenierizados, más o menos complejos, se encuentran actualmente disponibles en el mercado (Ehrenreich y Ruszczak, 2006Ehrenreich, M. y Ruszczak, Z. (2006). Update on tissue-engineered biological dressings. Tissue Engineering, 12 (9), pp. 2407-2424. https://doi.org/10.1089/ten.2006.12.2407.) y han sido empleados con éxito en la clínica para la regeneración tisular permanente en distintas patologías (Wong, McGrath y Navsaria, 2007Wong, T., McGrath, J. A. y Navsaria, H. (2007). The role of fibroblasts in tissue engineering and regeneration. British Journal of Dermatology, 156 (6), pp. 1149-1155. https://doi.org/10.1111/j.1365-2133.2007.07914.x.). El equivalente cutáneo ideal debería ser fácil de manipular, resistente y barato, además de ser capaz de recrear la fisiología de la piel y no inducir rechazo inmunológico en el receptor. Las matrices dérmicas de dichos sustitutos pueden estar compuestas de varios materiales, como colágeno de origen humano o bovino, proteoglicanos, ácido hialurónico o polímeros sintéticos, entre otros. Sin embargo, se ha comprobado que los queratinocitos crecidos sobre este tipo de matrices pierden capacidad de expansión. En los últimos años se ha conseguido optimizar este sistema mediante el desarrollo de una piel bioingenierizada compuesta por queratinocitos humanos que proliferan sobre una matriz de fibrina en la que se encuentran embebidos fibroblastos dérmicos humanos vivos. Este sustituto cutáneo presenta numerosas ventajas frente a los implantes de láminas epiteliales entre las que destacan la gran capacidad de expansión del componente epitelial in vitro y la preservación funcional de las CMEs. Esto es probablemente debido a que la fibrina, componente fundamental de la matriz provisional durante el proceso fisiológico de cicatrización, actúa como un fuerte inductor de la re-epitelización y la reorganización del tejido conectivo, eventos fundamentales para la regeneración cutánea. La fibrina no solo constituye un reservorio de diferentes factores sino que además preserva la potencialidad y funcionalidad de las CMEs, ya que favorece la adhesión de células indiferenciadas. Por todo ello, su uso está muy extendido para la regeneración de piel en casos de pérdidas masivas de tejido cutáneo (Llames et al., 2004Llames, S. G., Río, M. del, Larcher, F., García, E., García, M., Escámez, M. J. [...] y Meana, A. (2004). Human plasma as a dermal scaffold for the generation of a completely autologous bioengineered skin. Transplantation, 77 (3), pp. 350-355. https://doi.org/10.1097/01.TP.0000112381.80964.85.; Llames et al., 2006Llames, S., García, E., García, V., Río, M. del, Larcher, F., Jorcano, J. L. [...] y Meana, A. (2006). Clinical results of an autologous engineered skin. Cell and Tissue Banking, 7 (1), pp. 47-53. https://doi.org/10.1007/s10561-004-7253-4.) y en otros tejidos como el cardíaco, óseo o cartilaginoso, entre otros.
En ensayos preclínicos, la piel bioingenierizada puede ser trasplantada a ratones inmunodeficientes de manera que la piel humana regenerada retiene las principales características clínicas e histológicas de la piel humana madura (tanto en el nivel dérmico como en el epidérmico) y persiste durante toda la vida del animal. Cuando las células provienen de piel de pacientes, es posible recapitular el fenotipo de la enfermedad en la piel regenerada en el ratón. Dicho modelo es lo que se conoce como modelo de ratón humanizado en piel (Río et al., 2002Río, M. del, Larcher, F., Serrano, F., Meana, A., Muñoz, M., García, M. [...] y Jorcano, J. L. (2002). A preclinical model for the analysis of genetically modified human skin in vivo. Human Gene Therapy, 13 (8), pp. 959-968. https://doi.org/10.1089/10430340252939069.) y ha permitido estudiar la fisiopatología de diversas patologías cutáneas prevalentes y raras, así como ayudar al diseño y evaluación de estrategias de intervención terapéutica en el campo de la dermatología (Escámez et al., 2011Escámez, M. J., Martínez-Santamaría, L., García, M., Guerrero-Aspizua, S., Carretero, M., Larcher, F., Meana, A. y Río, M. del (2011). Bioengineered Skin. En: Khopkar, U. (ed.). Skin Biopsy. Perspectives. InTech, pp. 261-296.).
Las investigaciones en el campo de la IT se unen a los incesantes avances en el desarrollo de nuevas tecnologías como el bioprinting y el conocimiento de las potenciales propiedades regenerativas de las células madre (embrionarias y adultas). De hecho, la generación de sustitutos bioingenierizados a partir de células pluripotentes inducidas son también ya una realidad en el campo de IT cutánea (Guo et al., 2013Guo, Z., Higgins, C. A., Gillette, B. M., Itoh, M., Umegaki, N., Gledhill, K. [...] y Christiano, A. M. (2013). Building a microphysiological skin model from induced pluripotent stem cells. Stem Cell Research Therapy, 4 (1), S2. https://doi.org/10.1186/scrt363.; Lee et al, 2014Lee, V., Singh, G., Trasatti, J. P., Bjornsson, C., Xu, X., Tran, T. N. [...] y Karande, P. (2014). Design and fabrication of human skin by three-dimensional bioprinting. Tissue Engineering. Part C: Methods, 20 (6), pp. 473-484. https://doi.org/10.1089/ten.tec.2013.0335.).
TERAPIAS AVANZADAS PARA ENFERMEDADES RARAS DE LA PIEL Top
Las enfermedades hereditarias raras de piel, también conocidas como genodermatosis, representan una parte relevante de la patología dermatológica y constituyen cerca del 8% del conjunto de enfermedades raras. Actualmente, las bases genéticas de alrededor de 400 genodermatosis, en su mayoría monogénicas, han sido dilucidadas, lo cual permite una clasificación precisa, su diagnóstico molecular y, en buena medida, la comprensión del mecanismo patogénico. Sin embargo, en la inmensa mayoría de los casos no existe tratamiento curativo sino fundamentalmente paliativo, y a menudo las consecuencias de las genodermatosis son devastadoras y estigmatizantes para el paciente.
La epidermolisis bullosa (EB) es la primera familia de genodermatosis cuyo tratamiento ha sido abordado por terapia génica ex vivo. El tratamiento en 2006 de un paciente con CMEs autólogas modificadas genéticamente evidenció el potencial terapéutico de esta estrategia. El conocimiento acumulado desde entonces ha sido clave en el diseño de tratamientos para otras enfermedades monogénicas de la piel.
La EB es un grupo de enfermedades raras que se caracteriza por la presencia de erosiones y ampollas en la piel y mucosas, debidas a mutaciones en genes que codifican varias proteínas estructurales responsables de mantener la adhesión dermo-epidérmica.
Epidermolisis bullosa juntural (EBJ). La EBJ se debe a mutaciones en los genes que codifican para las cadenas α3, β3, y γ2 de la laminina 332, la integrina α6β4 o el colágeno XVII. La EBJ fue la primera genodermatosis que se abordó con terapia génica en un estudio llevado a cabo en Italia por el grupo liderado por los Dres. Michele de Luca y Fulvio Mavilio. Esta pionera aproximación terapéutica consistió en el trasplante de láminas de queratinocitos modificados genéticamente con vectores retrovirales convencionales para producir la cadena β3 de la laminina 332 (Mavilio et al., 2006Mavilio, F., Pellegrini, G., Ferrari, S., Nunzio, F. di, Iorio, E., Recchia, A. [...] y Luca, M. de (2006). Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nature Medicine, 12 (12), pp. 1397-1402. https://doi.org/10.1038/nm1504.). Este protocolo mostró beneficios terapéuticos indiscutibles. Es de destacar que, a los doce años del tratamiento, el paciente continúa expresando la proteína terapéutica y la piel regenerada sigue mostrando una buena resistencia mecánica (Carulli, Contin, Rosa, Pellegrini y Luca, 2013Carulli, S., Contin, R., Rosa, L. de, Pellegrini, G. y Luca, M. de (2013). The long and winding road that leads to a cure for epidermolysis bullosa. Regenerative Medicine, 8 (4), pp. 467-481. https://doi.org/10.2217/rme.13.33.; Rosa et al., 2013Rosa, L. de, Carulli, S., Cocchiarella, F., Quaglino, D., Enzo, E., Franchini, E. [...] y Luca, M. de (2013). Long-term stability and safety of transgenic cultured epidermal stem cells in gene therapy of junctional epidermolysis bullosa. Stem Cell Reports, 2 (1), pp. 1-8. https://doi.org/10.1016/j.stemcr.2013.11.001.). Utilizando el mismo protocolo, una nueva paciente fue trasplantada en Salzburgo en julio de 2014 por el grupo del Dr. Bauer en colaboración con el Dr. de Luca, mostrando una regeneración estable de la úlcera tratada tres meses y medio después del trasplante (Bauer et al., 2017Bauer, J. W., Koller, J., Murauer, E. M., Rosa, L., Enzo, E., Carulli, S. [...] y Luca, M. de (2017). Closure of a large chronic wound through transplantation of gene-corrected epidermal stem cells. The Journal of Investigative Dermatology, 137 (3), pp. 778-781. https://doi.org/10.1016/j.jid.2016.10.038.).
Epidermolisis bullosa distrófica (EBD). La EBD está causada por mutaciones en el gen COL7A1 que codifica el colágeno tipo VII (C7). El C7, principal componente de las fibrillas de anclaje, desempeña un papel crítico en la adhesión dermo-epidérmica y por tanto en el mantenimiento de la integridad cutánea. Los enfoques terapéuticos para EBD se centran en la restauración de la expresión o la reposición del C7 mediante abordajes de terapia génica ex vivo, terapia celular e ingeniería tisular (figura 2). Si bien los queratinocitos son la fuente natural y mayoritaria de C7, los fibroblastos dérmicos, aunque en menor grado, son igualmente capaces de secretar esta proteína. Por tanto, se está valorando la oportunidad de utilizar estrategias de terapia celular basadas en la administración local de fibroblastos alogénicos (células con mejor tolerancia inmunológica que los queratinocitos) (Petrof, Martínez-Queipo, Mellerio, Kemp y McGrath, 2015Petrof, G., Martínez-Queipo, M., Mellerio, J. E., Kemp, P. y McGrath, J. A. (2013). Fibroblast cell therapy enhances initial healing in recessive dystrophic epidermolysis bullosa wounds: results of a randomized, vehicle-controlled trial. British Journal of Dermatology, 169 (5), pp. 1025-1033. https://doi.org/10.1111/bjd.12599.; Venugopal et al., 2013Venugopal, S. S., Yan, W., Frew, J. W., Cohn, H. I., Rhodes, L. M., Tran, K. [...] y Murrell, D. F. (2013). A phase II randomized vehicle-controlled trial of intradermal allogeneic fibroblasts for recessive dystrophic epidermolysis bullosa. Journal of American Academy of Dermatology, 69 (6), pp. 898-908. https://doi.org/10.1016/j.jaad.2013.08.014.). Cabe destacar el ensayo realizado en Londres por la empresa Intercytex (Eudract 2010-023121-38) que, si bien obtuvo resultados modestos en los once pacientes tratados, demostró la posibilidad de emplear células de origen mesenquimal como fuente alternativa de C7, así como la ausencia de rechazo clínico (Petrof et al., 2013Petrof, G., Martínez-Queipo, M., Mellerio, J. E., Kemp, P. y McGrath, J. A. (2013). Fibroblast cell therapy enhances initial healing in recessive dystrophic epidermolysis bullosa wounds: results of a randomized, vehicle-controlled trial. British Journal of Dermatology, 169 (5), pp. 1025-1033. https://doi.org/10.1111/bjd.12599.).
|
Figura 2. Terapias avanzadas para la epidermolisis bullosa distrófica (EBD). Esquema de las diferentes estrategias terapéuticas en desarrollo
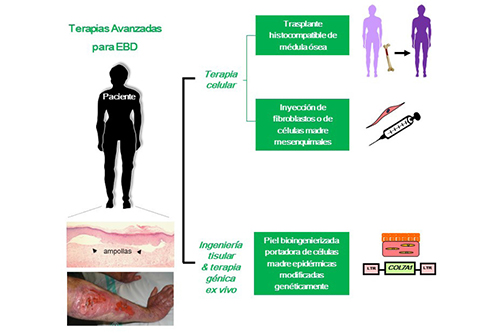
Fuente: Elaboración propia.
[Descargar tamaño completo]
| |
En la actualidad se están llevando a cabo varios ensayos clínicos de terapia génica ex vivo para EBD. En uno de ellos participa la Unidad 714 del CIBERER (UC3M-CIEMAT/IIS Fundación Jiménez Díaz) y está enmarcado en el proyecto europeo Genegraft. La estrategia se basa en evidencias preclínicas obtenidas diez años antes por la U714 en colaboración con el grupo del Dr. Meneguzzi en un modelo humanizado de EBD (Gache et al., 2004Gache, Y., Baldeschi, C., Río, M. del, Gagnoux-Palacios, L., Larcher, F., Lacour, J. P. y Meneguzzi, G. (2004). Construction of skin equivalents for gene therapy of recessive dystrophic epidermolysis bullosa. Human Gene Therapy, 15 (10), pp. 921-933. https://doi.org/10.1089/hum.2004.15.921.). Dicha aproximación consiste en el trasplante de CMEs modificadas genéticamente asociadas a una matriz dérmica que contiene fibroblastos. Este equivalente dermo-epidérmico, generado por ingeniería de tejidos, ha obtenido la designación de medicamento huérfano (EU/3/09/630) y será empleado en el ensayo europeo liderado por el Dr. Hovnanian (NCG01874769; Hovnanian, Bodemer y McGrath, 2014Hovnanian, A., Bodemer, C. y McGrath, J. A. (2014). Phase I/II ex vivo gene therapy clinical trial for recessive dystrophic epidermolysis bullosa using skin equivalent grafts genetically corrected with a COL7A1-encoding SIN retroviral vector (GENEGRAFT). Human Gene Therapy Clinical Development, 25 (2), pp. 65-66. https://doi.org/10.1089/humc.2014.2508.). El otro ensayo, liderado por el Dr. Lane, se está llevando en la Universidad de Stanford (NCT01263379) y se basa fundamentalmente en el mismo concepto. Ambos estudios están activos y reclutando pacientes. El grupo americano ha publicado resultados preliminares prometedores en un primer grupo de cuatro pacientes tratados (Siprashvili et al., 2016Siprashvili, Z., Nguyen, N. T., Gorell, E. S., Loutit, K., Khuu, P., Furukawa, L. K. [...] y Marinkovich, M. P. (2016). Safety and wound outcomes following genetically corrected autologous epidermal grafts in patients with recessive dystrophic epidermolysis bullosa. JAMA, 316 (17), pp. 1808-1817. https://doi.org/10.1001/jama.2016.15588.) y está reclutando nuevos pacientes para comenzar un ensayo clínico en fase II. Más recientemente y tras el éxito preclínico del transplante de piel bioingenierizada portadora de fibroblastos transducidos con un vector lentiviral auto-inactivador (Georgiadis et al., 2016Georgiadis, C., Syed, F., Petrova, A., Abdul-Wahab, A., Lwin, S. M., Farzaneh, F. [...] y Qasim, W. (2016). Lentiviral engineered fibroblasts expressing codon-optimized COL7A1 restore anchoring fibrils in RDEB. Journal of Investigative Dermatology, 136 (1), pp. 284-292. https://doi.org/10.1038/JID.2015.364.), se ha lanzado el primer ensayo clínico basado en la administración intradérmica de fibroblastos autólogos modificados genéticamente para el tratamiento de EBD recesiva (NCT02493816). En la actualidad está en marcha un ensayo clínico en fase I/II de terapia génica ex vivo con fibroblastos (NCT02810951) en pacientes adultos y en niños con EBD recesiva. Por otro lado, en el año 2010, y para sorpresa de parte de la comunidad científica, se publicaron los resultados de un estudio clínico para EBD que utilizó trasplante de medula ósea de hermanos histocompatibles. El estudio demostró una marcada mejoría clínica en seis de los siete pacientes enrolados (NCT00478244; Wagner et al., 2010Wagner, J. E., Ishida-Yamamoto, A., McGrath, J. A., Hordinsky, M., Keene, D. R., Woodley, D. T. [...] y Tolar, J. (2010). Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. The New England Journal of Medicine, 363 (7), pp. 629-639. https://doi.org/10.1056/NEJMoa0910501.). Concretamente, se observó depósito de C7 en la unión dermo-epidérmica, así como persistencia de las células del donante en la piel de los pacientes. Cabe destacar que, al tratarse de un abordaje sistémico, presentaba la ventaja añadida de mejorar también la fragilidad de epitelios internos. Sin embargo, el acondicionamiento de los pacientes, así como la incidencia de enfermedad injerto contra huésped, asociados al trasplante de médula ósea, cuestionaron su empleo en EBD. De hecho, en un estudio posterior en veinte pacientes, cinco pacientes fallecieron a causa de complicaciones asociadas al tratamiento (Tolar y Wagner, 2013Tolar, J. y Wagner, J. E. (2013). Allogeneic blood and bone marrow cells for the treatment of severe epidermolysis bullosa: repair of the extracellular matrix. The Lancet, 382 (9899), pp. 1214-1223. https://doi.org/10.1016/S0140-6736(13)61897-8.). No obstante, estos ensayos fueron un acontecimiento en la historia del tratamiento de la EBD y han abierto nuevas e interesantes líneas de investigación en el campo. Así, experimentos preclínicos recientes han demostrado que la población celular de la médula ósea con capacidad regenerativa en EBD es consistente con CMMs (Tamai et al., 2011Tamai, K., Yamazaki, T., Chino, T., Ishii, M., Otsuru, S., Kikuchi, Y. [...] y Kaneda, Y. (2011). PDGFRalpha-positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proceedings of the National Academy of Sciences USA, 108 (16), pp. 6609-6614. https://doi.org/10.1073/pnas.1016753108.). Estos estudios, así como los resultados beneficiosos obtenidos en dos pacientes en los que además se observó una ausencia de efectos adversos relevantes (Conget et al., 2010Conget, P., Rodríguez, F., Kramer, S., Allers, C., Simon, V., Palisson, F. [...] y Yubero, M. J. (2010). Replenishment of type VII collagen and re-epithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy, 12 (3), pp. 429-431. https://doi.org/10.3109/14653241003587637.), han dado lugar a la puesta en marcha de tres ensayos clínicos. Uno de ellos se está realizando en Japón y consiste en la administración intradérmica de CMMs derivadas de médula ósea (UMIN000006723). En cuanto a los otros dos ensayos clínicos, basados en la administración sistémica de CMMs, se han realizado en el Reino Unido (SRCTN46615946) y en Egipto. En ambos casos se ha comprobado una mejoría en la resistencia cutánea en los pacientes tratados (El-Darouti et al., 2016El-Darouti, M., Fawzy, M., Amin, I., Abdel Hay, R., Hegazy, R., Gabr, H. y El-Maadawi, Z. (2016). Treatment of dystrophic epidermolysis bullosa with bone marrow non-hematopoeitic stem cells: a randomized controlled trial. Dermatologic Therapy, 29 (2), pp. 96-100. https://doi.org/10.1111/dth.12305.; Petrof et al., 2015Petrof, G., Lwin, S. M., Martínez-Queipo, M., Abdul-Wahab, A., Tso, S., Mellerio, J. E. [...] y McGrath, J. A. (2015). Potential of systemic allogeneic mesenchymal stromal cell therapy for children with recessive dystrophic epidermolysis bullosa. The Journal of Investigative Dermatology, 135 (9), pp. 2319-2321. https://doi.org/10.1038/jid.2015.158.). En la actualidad se encuentran abiertos dos nuevos ensayos clínicos que utilizarán CMMs, uno de ellos en población adulta en Londres (NCT02323789) y otro en pacientes pediátricos en España (código EudraCT: 2017-000606-37), en el cual participa la Unidad 714 del CIBERER (UC3MCIEMAT/IIS Fundación Jiménez Díaz).
Síndrome de Netherton (SN). El SN está causado por mutaciones en el gen SPINK5 que codifica la proteína LEKTI, clave en el mantenimiento fisiológico de la función barrera de la piel. La falta de LEKTI da lugar a infecciones recurrentes, deshidratación, así como complicaciones tales como bronconeumonía y sepsis. Estos pacientes presentan mal pronóstico con una alta tasa de mortalidad (10% durante el primer año de vida). Los doctores Li y Qasim están en la fase inicial de un ensayo clínico de terapia génica ex vivo en el hospital Great Ormond Street de Londres (NCT01545323). Este ensayo se basa en resultados preclínicos obtenidos en colaboración con la U714, en los que se demostró que la transferencia del gen terapéutico a las CMEs mediante vectores lentivirales era capaz de restablecer un estrato córneo funcional (Di et al., 2011Di, W. L., Larcher, F., Semenova, E., Talbot, G. E., Harper, J. I., Río, M. del [...] y Qasim, W. (2011). Ex-vivo gene therapy restores LEKTI activity and corrects the architecture of Netherton syndrome-derived skin grafts. Molecular Therapy, 19 (2), pp. 408-416. https://doi.org/10.1038/mt.2010.201.).
Paquioniquia congénita (PC). A diferencia de las genodermatosis comentadas anteriormente, que tienen una herencia autosómica recesiva, la PC es un desorden de la queratinización que presenta una herencia autosómica dominante. La enfermedad se caracteriza por la aparición de queratodermia palmoplantar sumamente dolorosa e incapacitante para los pacientes. La PC está causada por mutaciones en los genes que codifican para las keratinas K6, K16 y K17. El grupo del Dr. Kaspar en EEUU diseñó un siARN (small interfering ARN/ARN de interferencia) capaz de anular específicamente la función del gen mutado (terapia génica de supresión) sin alterar el producto del alelo normal. Este equipo demostró además la viabilidad de la estrategia en estudios in vitro (Hickerson et al., 2008Hickerson, R. P., Smith, F. J., Reeves, R. E., Contag, C. H., Leake, D., Leachman, S. A. [...] y Kaspar, R. L. (2008). Single-nucleotide-specific siARN targeting in a dominant-negative skin model. Journal of Investigative Dermatology, 128 (3), pp. 594-605. https://doi.org/10.1038/sj.jid.5701060.). En 2008 se llevó a cabo un ensayo clínico empleando siARN como tratamiento in vivo de la PC (NCT00716014), siendo el primer protocolo que utilizó esta tecnología en humanos. Este ensayo de fase Ib se realizó en una única paciente y demostró, por un lado, una clara mejoría clínica pero, por otro, la necesidad de mejorar el sistema de administración y su eficacia (Leachman et al., 2010Leachman, S. A., Hickerson, R. P., Schwartz, M. E., Bullough, E. E., Hutcherson, S. L., Boucher, K. M. [...] y Kaspar, R. L. (2010). First-in-human mutation-targeted siARN phase Ib trial of an inherited skin disorder. Molecular Therapy, 18 (2), pp. 442-446. https://doi.org/10.1038/mt.2009.273.). Un nuevo ensayo clínico se está preparando en la actualidad en el que participarán el Dr. Milstone (Universidad de Yale) y el Dr. Irvine (Our Lady’s Hospital, Dublín), entre otros.
RETOS Y OPORTUNIDADES Top
Las terapias avanzadas para el tratamiento de las ER se encuentran en un momento de expansión. En el caso concreto de la terapia génica han sido necesarios más de 20 años entre el primer ensayo clínico y el primer medicamento comercializado; sin embargo, en pocos años probablemente tengamos una imagen bien distinta. El número de medicamentos huérfanos basados en terapias avanzadas y el número de ensayos clínicos en desarrollo avanza significativamente cada año.
Por otro lado, es importante señalar las oportunidades que las nuevas tecnologías están aportando al campo. En este sentido destacamos los avances en la denominada cirugía o edición genética que empezaron con la aplicación de las nucleasas de dedos de zinc (ZFN), las secuencias TALEN, y que actualmente se sirven de la revolución que está suponiendo el sistema CRISPR/Cas9 para la edición del genoma (Lombardo y Naldini, 2014Lombardo, A. y Naldini, L. (2014). Genome Editing: A tool for research and therapy: Targeted genome editing hits the clinic. Nature Medicine, 20 (10), pp. 1101-1103. https://doi.org/10.1038/nm.3721.). La combinación de esta tecnología con el empleo de células madre pluripotentes inducidas diferenciadas a la estirpe celular deseada abre un gran campo de futuro, todavía hoy en fase experimental.
En definitiva, pues, estamos ante un escenario en que las aproximaciones más clásicas en terapias avanzadas están implantándose en ensayos clínicos, a la vez que irrumpen con fuerza nuevas tecnologías que previsiblemente irán a mejorar la seguridad y eficacia de estas terapias. Paralelamente el impulso que las nuevas metodologías en secuenciación del exoma y genoma están aportando a la identificación de las causas genéticas de enfermedades raras amplía el espectro de enfermedades cuya opción terapéutica contemple tratamientos de terapias avanzadas.
AGRADECIMIENTOSTop
Los laboratorios de los autores están financiados por: Laboratorio IDIBAPS/CIBERER, Ministerio de Economía y Competitividad (BIO2014-57716-C2-2-R), Instituto de Salud Carlos III (IIS10/00014), Generalitat de Catalunya (SGR14/248), Fondation Jerome Lejeune (FJL14). El trabajo realizado por el grupo del CIEMAT/CIBERER/IIS Fundación Jiménez Díaz (UAM) está financiado por la Comisión Europea (FP7 HEALTH-F5-2012-305421, EUROFANCOLEN), Ministerio de Economía y Competitividad (SAF2012-39834), Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III (RETICS RD12/0019/0023; PI08/0701), y Dirección General de Investigación de la Comunidad de Madrid (CellCAM; Ref S2010/BMD-2420).
BIBLIOGRAFÍATop
|
| ○ | Adair, J. E., Sevilla, J., Díaz de Heredia, C., Becker, P. S., Kiem, H-P. y Bueren, J. A. (2016). Lessons Learned from Two Decades of Clinical Trial Experience in Gene Therapy for Fanconi Anemia. Current Gene Therapy, 16 (5), pp. 338-348. https://doi.org/10.2174/1566523217666170119113029 |
| ○ | Aiuti, A., Biasco, L., Scaramuzza, S., Ferrua, F., Cicalese, M. P., Baricordi, L. [...] y Naldini, L. (2013). Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science, 341 (6148), 1233151. https://doi.org/10.1126/science.1233151 |
| ○ | Aiuti, A. y Roncarolo, M. G. (2009). Ten years of gene therapy for primary immune deficiencies. Hematology. American Society of Hematology Education Program Book, 2009 (1), pp. 682-689. |
| ○ | Aiuti, A., Roncarolo, M. G. y Naldini, L. (2017). Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: paving the road for the next generation of advanced therapy medicinal products. EMBO Molecular Medicine, 9 (6), e201707573. https://doi.org/10.15252/emmm.201707573 |
| ○ | Aiuti, A., Slavin, S., Aker, M., Ficara, F., Deola, S., Mortellaro, A. [...] y Bordignon, C. (2002). Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science, 296 (5577), pp. 2410-2413. https://doi.org/10.1126/science.1070104 |
| ○ | Atala, A. (2007). Engineering tissues, organs and cells. Journal of Tissue Engineering and Regenerative. Medicine, 1 (2), pp. 83-96. https://doi.org/10.1002/term.18 |
| ○ | Bainbridge, J. W., Smith, A. J., Barker, S. S, Robbie, S., Henderson, R., Balaggan, K. [...] y Ali, R. R. (2008). Effect of gene therapy on visual function in Leber’s congenital amaurosis. The New England Journal of Medicine, 358 (21), pp. 2231-2239. https://doi.org/10.1056/NEJMoa0802268 |
| ○ | Bajaj, P., Schweller, R. M., Khademhosseini, A., West, J. L. y Bashir, R. (2014). 3D biofabrication strategies for tissue engineering and regenerative medicine. Annual Review of Biomedical Engineering, 16, pp. 247-276. https://doi.org/10.1146/annurev-bioeng-071813-105155 |
| ○ | Bauer, J. W., Koller, J., Murauer, E. M., Rosa, L., Enzo, E., Carulli, S. [...] y Luca, M. de (2017). Closure of a large chronic wound through transplantation of gene-corrected epidermal stem cells. The Journal of Investigative Dermatology, 137 (3), pp. 778-781. https://doi.org/10.1016/j.jid.2016.10.038 |
| ○ | Bennett, J., Ashtari, M., Wellman, J., Marshall, K. A., Cyckowski, L. L., Chung, D. C. [...] y Maguire, A. M. (2012). AAV2 gene therapy readministration in three adults with congenital blindness. Science Translational Medicine, 4 (120), 120ra15. https://doi.org/10.1126/scitranslmed.3002865 |
| ○ | Biffi, A., Montini, E., Lorioli, L., Cesani, M., Fumagalli, F., Plati, T. [...] y Naldini, L. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science, 341 (6148), 1233158. https://doi.org/10.1126/science.1233158 |
| ○ | Blaese, R. M., Culver, K. W., Miller, A. D., Carter, C. S., Fleisher, T., Clerici, M., Shearer, G. [...] y Anderson, W. F. (1995). T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science, 270 (5235), pp. 475-480. https://doi.org/10.1126/science.270.5235.475 |
| ○ | Bordignon, C., Mavilio, F., Ferrari, G., Servida, P., Ugazio, A. G., Notarangelo, L. D. [...] y Gillio, A. P. (1993). Transfer of the ADA gene into bone marrow cells and peripheral blood lymphocytes for the treatment of patients affected by ADA-deficient SCID. Human Gene Therapy, 4 (4), pp. 513-520. https://doi.org/10.1089/hum.1993.4.4-513 |
| ○ | Boztug, K., Schmidt, M., Schwarzer, A., Banerjee, P. P., Díez, I. A., Dewey, R. A. [...] y Klein, C. (2010). Stem-cell gene therapy for the Wiskott-Aldrich syndrome. The New England Journal of Medicine, 363 (20), pp. 1918-1927. https://doi.org/10.1056/NEJMoa1003548 |
| ○ | Braun, C. J., Boztug, K., Paruzynski, A., Witzel, M., Schwarzer, A., Rothe, M. [...] y Klein, C. (2014). Gene therapy for Wiskott-Aldrich syndrome--long-term efficacy and genotoxicity. Science Transational. Medicine, 6 (227), 227ra33. https://doi.org/10.1126/scitranslmed.3007280 |
| ○ | Cartier, N., Hacein-Bey-Abina, S., Bartholomae, C. C., Veres, G., Schmidt, M., Kutschera, I. [...] y Aubourg, P. (2009). Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science, 326 (5954), pp. 818-823. https://doi.org/10.1126/science.1171242 |
| ○ | Carulli, S., Contin, R., Rosa, L. de, Pellegrini, G. y Luca, M. de (2013). The long and winding road that leads to a cure for epidermolysis bullosa. Regenerative Medicine, 8 (4), pp. 467-481. https://doi.org/10.2217/rme.13.33 |
| ○ | Cavazzana-Calvo, M., Hacein-Bey, S., Saint Basile, G. de, Gross, F., Yvon, E., Nusbaum, P. [...] y Fischer, A. (2000). Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science, 288 (5466), pp. 669-672. https://doi.org/10.1126/science.288.5466.669 |
| ○ | Cavazzana-Calvo, M., Payen, E., Negre, O., Wang, G., Hehir, K., Fusil, F. [...] y Leboulch, P. (2010). Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature, 467 (7313), pp. 318-322. |
| ○ | Conget, P., Rodríguez, F., Kramer, S., Allers, C., Simon, V., Palisson, F. [...] y Yubero, M. J. (2010). Replenishment of type VII collagen and re-epithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy, 12 (3), pp. 429-431. https://doi.org/10.3109/14653241003587637 |
| ○ | Di, W. L., Larcher, F., Semenova, E., Talbot, G. E., Harper, J. I., Río, M. del [...] y Qasim, W. (2011). Ex-vivo gene therapy restores LEKTI activity and corrects the architecture of Netherton syndrome-derived skin grafts. Molecular Therapy, 19 (2), pp. 408-416. https://doi.org/10.1038/mt.2010.201 |
| ○ | Ehrenreich, M. y Ruszczak, Z. (2006). Update on tissue-engineered biological dressings. Tissue Engineering, 12 (9), pp. 2407-2424. https://doi.org/10.1089/ten.2006.12.2407 |
| ○ | El-Darouti, M., Fawzy, M., Amin, I., Abdel Hay, R., Hegazy, R., Gabr, H. y El-Maadawi, Z. (2016). Treatment of dystrophic epidermolysis bullosa with bone marrow non-hematopoeitic stem cells: a randomized controlled trial. Dermatologic Therapy, 29 (2), pp. 96-100. https://doi.org/10.1111/dth.12305 |
| ○ | Escámez, M. J., Martínez-Santamaría, L., García, M., Guerrero-Aspizua, S., Carretero, M., Larcher, F., Meana, A. y Río, M. del (2011). Bioengineered Skin. En: Khopkar, U. (ed.). Skin Biopsy. Perspectives. InTech, pp. 261-296. |
| ○ | Fillat, C. y Altafaj, X. (2012). Gene Therapy for Down Syndrome. Progress in Brain Research, 197, pp. 237-247. https://doi.org/10.1016/B978-0-444-54299-1.00012-1 |
| ○ | Gache, Y., Baldeschi, C., Río, M. del, Gagnoux-Palacios, L., Larcher, F., Lacour, J. P. y Meneguzzi, G. (2004). Construction of skin equivalents for gene therapy of recessive dystrophic epidermolysis bullosa. Human Gene Therapy, 15 (10), pp. 921-933. https://doi.org/10.1089/hum.2004.15.921 |
| ○ | García-Gómez, M., Calabria, A., García-Bravo, M., Benedicenti, F., Kosinski, P., López-Manzaneda, S. [...] y Segovia, J. C. (2016). Safe and Efficient Gene Therapy for Pyruvate Kinase Deficiency. Molecular Therapy, 24 (7), pp. 1187-1198. https://doi.org/10.1038/mt.2016.87 |
| ○ | Gaspar, H. B., Parsley, K. L., Howe, S., King, D., Gilmour, K. C., Sinclair, J. [...] y Thrasher, A. J. (2004). Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. The Lancet, 364 (9452), pp. 2181-2187. https://doi.org/10.1016/S0140-6736(04)17590-9 |
| ○ | Georgiadis, C., Syed, F., Petrova, A., Abdul-Wahab, A., Lwin, S. M., Farzaneh, F. [...] y Qasim, W. (2016). Lentiviral engineered fibroblasts expressing codon-optimized COL7A1 restore anchoring fibrils in RDEB. Journal of Investigative Dermatology, 136 (1), pp. 284-292. https://doi.org/10.1038/JID.2015.364 |
| ○ | González-Murillo, A., Lozano, M. L., Álvarez, L., Jacome, A., Almarza, E., Navarro, S. [...] y Río, P. (2010). Development of lentiviral vectors with optimized transcriptional activity for the gene therapy of patients with Fanconi anemia. Human Gene Therapy, 21 (5), pp. 623-630. https://doi.org/10.1089/hum.2009.141 |
| ○ | Guo, Z., Higgins, C. A., Gillette, B. M., Itoh, M., Umegaki, N., Gledhill, K. [...] y Christiano, A. M. (2013). Building a microphysiological skin model from induced pluripotent stem cells. Stem Cell Research Therapy, 4 (1), S2. https://doi.org/10.1186/scrt363 |
| ○ | Hacein-Bey-Abina, S., Deist, F. Le, Carlier, F., Bouneaud, C., Hue, C., Villartay, J. P. de [...] y Cavazzana-Calvo, M. (2002). Sustained correction of X-Linked severe combined immunodeficiency by ex vivo gene therapy. The New England Journal of Medicine, 346 (16), pp. 1185-1193. https://doi.org/10.1056/NEJMoa012616 |
| ○ | Hacein-Bey-Abina, S., Pai, S. Y., Gaspar, H. B., Armant, M., Berry, C. C., Blanche, S. [...] y Thrasher, A. J. (2014). A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. The New England Journal of Medicine, 371 (15), pp. 1407-1417. https://doi.org/10.1056/NEJMoa1404588 |
| ○ | Hacein-Bey-Abina, S., Saint Basile, G. de y Cavazzana-Calvo, M. (2003). Gene therapy of X-linked severe combined immunodeficiency. Methods in Molecular Biology, 215, pp. 247-259. |
| ○ | Haurigot, V., Marcó, S., Ribera, A., García, M., Ruzo, A., Villacampa, P. [...] y Bosch, F. (2013). Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. The Journal of Clinical Investigation, 123 (8), pp. 3254-3271. https://doi.org/10.1172/JCI66778 |
| ○ | Hauswirth, W. W., Aleman, T. S., Kaushal, S., Cideciyan, A. V., Schwartz, S. B., Wang, L. [...] y Jacobson, S. G. (2008). Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Human Gene Therapy, 19 (10), pp. 979-990. https://doi.org/10.1089/hum.2008.107 |
| ○ | Hickerson, R. P., Smith, F. J., Reeves, R. E., Contag, C. H., Leake, D., Leachman, S. A. [...] y Kaspar, R. L. (2008). Single-nucleotide-specific siARN targeting in a dominant-negative skin model. Journal of Investigative Dermatology, 128 (3), pp. 594-605. https://doi.org/10.1038/sj.jid.5701060 |
| ○ | Hovnanian, A., Bodemer, C. y McGrath, J. A. (2014). Phase I/II ex vivo gene therapy clinical trial for recessive dystrophic epidermolysis bullosa using skin equivalent grafts genetically corrected with a COL7A1-encoding SIN retroviral vector (GENEGRAFT). Human Gene Therapy Clinical Development, 25 (2), pp. 65-66. https://doi.org/10.1089/humc.2014.2508 |
| ○ | Kelly, P. F., Radtke, S., Kalle, C. von, Balcik, B., Bohn, K., Mueller, R. [...] y Williams, D. A. (2007). Stem cell collection and gene transfer in Fanconi anemia. Molecular Therapy, 15 (1), pp. 211-219. https://doi.org/10.1038/sj.mt.6300033 |
| ○ | Kohn, D. B., Hershfield, M. S., Carbonaro, D., Shigeoka, A., Brooks, J., Smogorzewska, E. M. [...] y Parkman, R. (1998). T lymphocytes with a normal ADA gene accumulate after transplantation of transduced autologous umbilical cord blood CD34+ cells in ADA-deficient SCID neonates. Nature Medicine, 4 (7), pp. 775-780. https://doi.org/10.1038/nm0798-775 |
| ○ | Leachman, S. A., Hickerson, R. P., Schwartz, M. E., Bullough, E. E., Hutcherson, S. L., Boucher, K. M. [...] y Kaspar, R. L. (2010). First-in-human mutation-targeted siARN phase Ib trial of an inherited skin disorder. Molecular Therapy, 18 (2), pp. 442-446. https://doi.org/10.1038/mt.2009.273 |
| ○ | Lee, V., Singh, G., Trasatti, J. P., Bjornsson, C., Xu, X., Tran, T. N. [...] y Karande, P. (2014). Design and fabrication of human skin by three-dimensional bioprinting. Tissue Engineering. Part C: Methods, 20 (6), pp. 473-484. https://doi.org/10.1089/ten.tec.2013.0335 |
| ○ | León-Rico, D., Aldea, M., Sánchez-Baltasar, R., Mesa-Núñez, C., Record, J., Burns, S. O. [...] y Almarza, A. (2016). Lentiviral vector mediated correction of a mouse model of Leukocyte Adhesion Deficiency Type I. Human Gene Therapy, 27 (9), pp. 668-678. https://doi.org/10.1089/hum.2016.016 |
| ○ | Lombardo, A. y Naldini, L. (2014). Genome Editing: A tool for research and therapy: Targeted genome editing hits the clinic. Nature Medicine, 20 (10), pp. 1101-1103. https://doi.org/10.1038/nm.3721 |
| ○ | Llames, S., García, E., García, V., Río, M. del, Larcher, F., Jorcano, J. L. [...] y Meana, A. (2006). Clinical results of an autologous engineered skin. Cell and Tissue Banking, 7 (1), pp. 47-53. https://doi.org/10.1007/s10561-004-7253-4 |
| ○ | Llames, S. G., Río, M. del, Larcher, F., García, E., García, M., Escámez, M. J. [...] y Meana, A. (2004). Human plasma as a dermal scaffold for the generation of a completely autologous bioengineered skin. Transplantation, 77 (3), pp. 350-355. https://doi.org/10.1097/01.TP.0000112381.80964.85 |
| ○ | Maguire, A. M., Simonelli, F., Pierce, E. A., Pugh Jr., E. N, Mingozzi, F., Bennicelli, J. [...] y Bennett, J. (2008). Safety and efficacy of gene transfer for Leber’s congenital amaurosis. The New England Journal Medicine, 358 (21), pp. 2240-2248. https://doi.org/10.1056/NEJMoa0802315 |
| ○ | Mavilio, F., Pellegrini, G., Ferrari, S., Nunzio, F. di, Iorio, E., Recchia, A. [...] y Luca, M. de (2006). Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nature Medicine, 12 (12), pp. 1397-1402. https://doi.org/10.1038/nm1504 |
| ○ | Meza, N. W., Alonso-Ferrero, M. E., Navarro, S., Quintana-Bustamante, O., Valeri, A., García-Gómez, M. [...] y Segovia, J. C. (2009). Rescue of pyruvate kinase deficiency in mice by gene therapy using the human isoenzyme. Molecular Therapy, 17 (12), pp. 2000-2009. https://doi.org/10.1038/mt.2009.200 |
| ○ | Mingozzi, F. y High, K. A. (2011). Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nature Reviews Genetics, 12 (5), pp. 341-355. https://doi.org/10.1038/nrg2988 |
| ○ | Modlich, U., Navarro, S., Zychlinski, D., Maetzig, T., Knoess, S., Brugman, M. H. [...] y Baum, C. (2009). Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Molecular Therapy, 17 (11), pp. 1919-1928. https://doi.org/10.1038/mt.2009.179 |
| ○ | Ott, M. G., Schmidt, M., Schwarzwaelder, K., Stein, S., Siler, U., Koehl, U. [...] y Grez, M. (2006). Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nature Medicine, 12 (4), pp. 401-409. https://doi.org/10.1038/nm1393 |
| ○ | Petrof, G., Lwin, S. M., Martínez-Queipo, M., Abdul-Wahab, A., Tso, S., Mellerio, J. E. [...] y McGrath, J. A. (2015). Potential of systemic allogeneic mesenchymal stromal cell therapy for children with recessive dystrophic epidermolysis bullosa. The Journal of Investigative Dermatology, 135 (9), pp. 2319-2321. https://doi.org/10.1038/jid.2015.158 |
| ○ | Petrof, G., Martínez-Queipo, M., Mellerio, J. E., Kemp, P. y McGrath, J. A. (2013). Fibroblast cell therapy enhances initial healing in recessive dystrophic epidermolysis bullosa wounds: results of a randomized, vehicle-controlled trial. British Journal of Dermatology, 169 (5), pp. 1025-1033. https://doi.org/10.1111/bjd.12599 |
| ○ | Río, M. del, Larcher, F., Serrano, F., Meana, A., Muñoz, M., García, M. [...] y Jorcano, J. L. (2002). A preclinical model for the analysis of genetically modified human skin in vivo. Human Gene Therapy, 13 (8), pp. 959-968. https://doi.org/10.1089/10430340252939069 |
| ○ | Río, P., Navarro, S., Guenechea, G., Sánchez-Domínguez, R., Lamana, M. L., Yañez, R. [...] y Bueren, J. (2017). Engraftment and in vivo proliferation advantage of gene corrected mobilized CD34+ cells from Fanconi anemia patients. Blood, 130 (13), pp. 1535-1542. https://doi.org/10.1182/blood-2017-03-774174 |
| ○ | Rosa, L. de, Carulli, S., Cocchiarella, F., Quaglino, D., Enzo, E., Franchini, E. [...] y Luca, M. de (2013). Long-term stability and safety of transgenic cultured epidermal stem cells in gene therapy of junctional epidermolysis bullosa. Stem Cell Reports, 2 (1), pp. 1-8. https://doi.org/10.1016/j.stemcr.2013.11.001 |
| ○ | Salido, E., Rodríguez-Pena, M., Santana, A., Beattie, S. G., Petry, H. y Torres, A. (2011). Phenotypic correction of a mouse model for primary hyperoxaluria with adeno-associated virus gene transfer. Molecular Therapy, 19 (5), pp. 870-875. https://doi.org/10.1038/mt.2010.270 |
| ○ | Schröder, A. R., Shinn, P., Chen, H., Berry, C., Ecker, J. R. y Bushman, F. (2002). HIV-1 integration in the human genome favors active genes and local hotspots. Cell, 110 (4), pp. 521-529. https://doi.org/10.1016/S0092-8674(02)00864-4 |
| ○ | Siprashvili, Z., Nguyen, N. T., Gorell, E. S., Loutit, K., Khuu, P., Furukawa, L. K. [...] y Marinkovich, M. P. (2016). Safety and wound outcomes following genetically corrected autologous epidermal grafts in patients with recessive dystrophic epidermolysis bullosa. JAMA, 316 (17), pp. 1808-1817. https://doi.org/10.1001/jama.2016.15588 |
| ○ | Song, J. J. y Ott, H. C. (2011). Organ engineering based on decellularized matrix scaffolds. Trends in Molecular Medicine, 17 (8), pp. 424-432. https://doi.org/10.1016/j.molmed.2011.03.005 |
| ○ | Stein, S., Ott, M. G., Schultze-Strasser, S., Jauch, A., Burwinkel, B., Kinner, A. [...] y Grez, M. (2010). Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nature Medicine, 16 (2), pp. 198-204. https://doi.org/10.1038/nm.2088 |
| ○ | Tamai, K., Yamazaki, T., Chino, T., Ishii, M., Otsuru, S., Kikuchi, Y. [...] y Kaneda, Y. (2011). PDGFRalpha-positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proceedings of the National Academy of Sciences USA, 108 (16), pp. 6609-6614. https://doi.org/10.1073/pnas.1016753108 |
| ○ | Tolar, J., Becker, P. S., Clapp, D. W., Hanenberg, H., Heredia, C. D. de, Kiem, H. P. [...] y Bueren, J. (2012). Gene therapy for Fanconi anemia: one step closer to the clinic. Human Gene Therapy, 23 (2), pp. 141-144. https://doi.org/10.1089/hum.2011.237 |
| ○ | Tolar, J. y Wagner, J. E. (2013). Allogeneic blood and bone marrow cells for the treatment of severe epidermolysis bullosa: repair of the extracellular matrix. The Lancet, 382 (9899), pp. 1214-1223. https://doi.org/10.1016/S0140-6736(13)61897-8 |
| ○ | Torres-Torronteras, J., Viscomi, C., Cabrera-Pérez, R., Cámara, Y., Meo, I. di, Barquinero, J. [...] y Martí, R. (2014). Gene therapy using a liver-targeted AAV vector restores nucleoside and nucleotide homeostasis in a murine model of MNGIE. Molecular Therapy, 22 (5), pp. 901-907. https://doi.org/10.1038/mt.2014.6 |
| ○ | Venugopal, S. S., Yan, W., Frew, J. W., Cohn, H. I., Rhodes, L. M., Tran, K. [...] y Murrell, D. F. (2013). A phase II randomized vehicle-controlled trial of intradermal allogeneic fibroblasts for recessive dystrophic epidermolysis bullosa. Journal of American Academy of Dermatology, 69 (6), pp. 898-908. https://doi.org/10.1016/j.jaad.2013.08.014 |
| ○ | Wagner, J. E., Ishida-Yamamoto, A., McGrath, J. A., Hordinsky, M., Keene, D. R., Woodley, D. T. [...] y Tolar, J. (2010). Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. The New England Journal of Medicine, 363 (7), pp. 629-639. https://doi.org/10.1056/NEJMoa0910501 |
| ○ | Wong, T., McGrath, J. A. y Navsaria, H. (2007). The role of fibroblasts in tissue engineering and regeneration. British Journal of Dermatology, 156 (6), pp. 1149-1155. https://doi.org/10.1111/j.1365-2133.2007.07914.x |
|